Clear Sky Science · ja
線形テンソル化四角注意機構によりバイオ分子力場のためのスケーラブルで量子精度の基盤モデル
なぜより高速な分子ムービーが重要なのか
現代の創薬や材料設計は、分子がねじれ、曲がり、反応する様子をコンピュータ上で“ムービー”として再現することにますます依存しています。これらのシミュレーションは、薬剤がタンパク質のポケットにどう収まるかや、新素材が応力下でどう振る舞うかを明らかにします。しかし現在のツールは科学者に選択を迫ります:精度を犠牲にして高速化した手法か、現実的な複雑系には遅すぎる超高精度の量子計算か。本論文はLiTENとその力場モデルLiTEN-FFを紹介し、日常的な分子モデリングに適した速度で量子レベルの精度を達成することを目指す新しい人工知能アプローチを提示します。
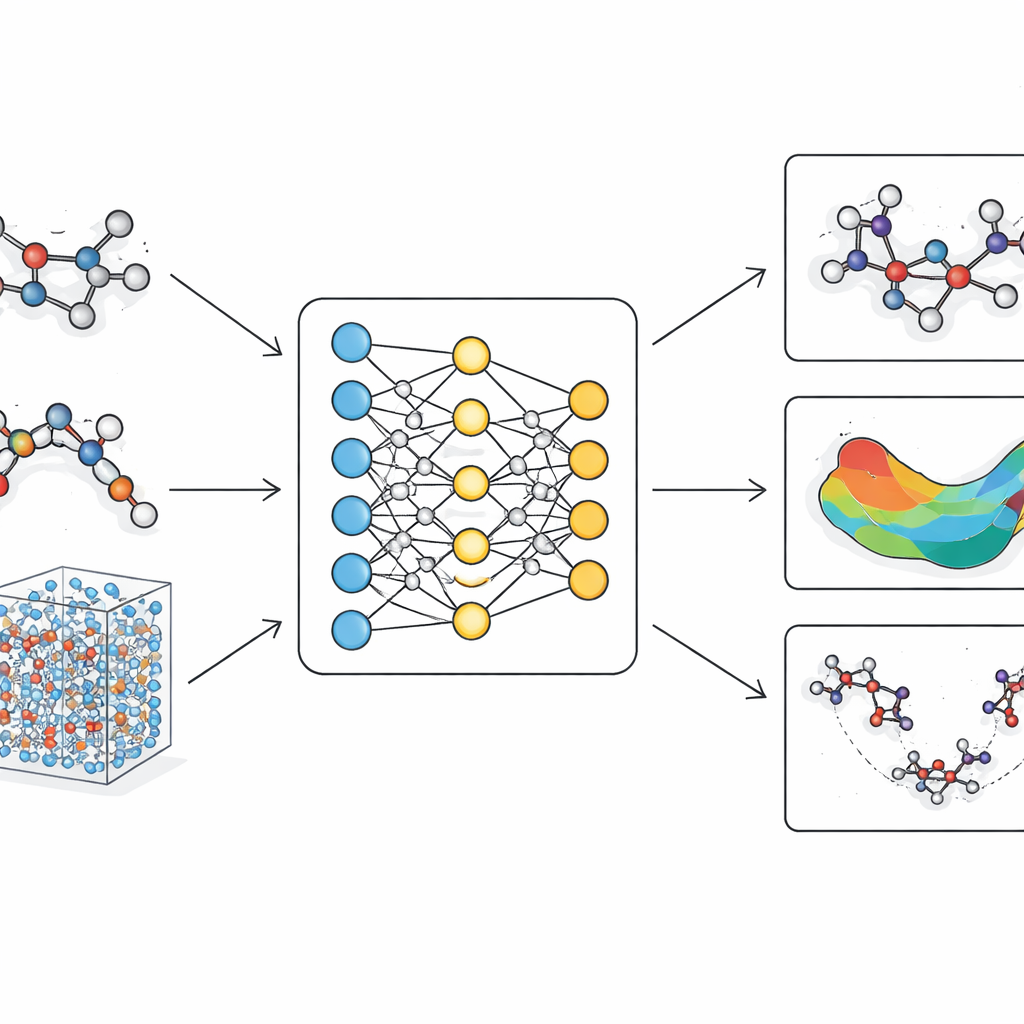
現在の分子モデルが抱える問題
従来の分子シミュレーションは大きく二つに分かれます。古典的力場は原子をスプリングで結ばれたビーズとして扱い固定パラメータを用います。これらは高速で大きなタンパク質や長時間スケールを扱えますが、微妙な形状変化、結合の再配置、化学反応経路といった実際の化学で重要な現象に弱い。一方、量子手法は電子を明示的に記述し、結合の切断・生成を正確に捉えますが、計算負荷が非常に大きいため通常は小さな分子や極めて短いシミュレーションに限定されます。近年では、量子計算を模倣することを学習する機械学習アプローチが中間解として登場しましたが、多くは信頼性に必要な物理的厳密性が欠けるか、現実的な大規模バイオ分子に拡張すると遅くなりすぎます。
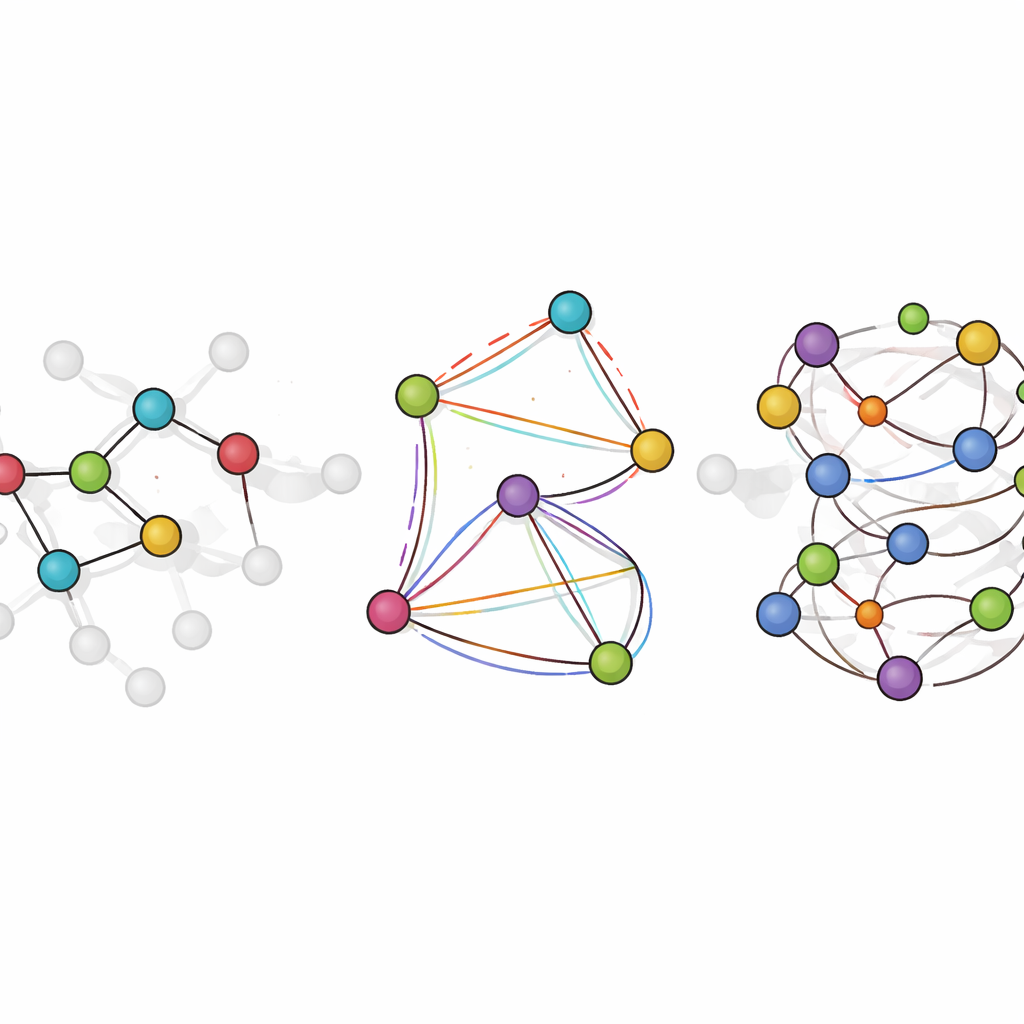
分子の形状をAIに教える新しい方法
LiTENはニューラルネットワークが分子幾何を“感じる”方法を再設計することでこの課題に取り組みます。原子間の単純な二体距離だけでなく、分子の角度やねじれを支配する三体・四体のパターン情報を組み込みます。重要なのは、分子を空間で回転させたり平行移動しても予測されるエネルギーは不変で、力は適切に回転するという基本的な物理対称性を保つ点です。鍵となる革新はテンソル化四角注意(tensorized quadrangle attention)と呼ばれる手法で、複雑な曲げやねじれの相互作用を、重い特殊数学ではなく効率的なベクトル演算で捉えます。これにより計算がスケーラブルになり、通常は高度なモデルを遅くする多体効果も系の大きさに対して線形にしかコストが増えない形で扱えます。
量子データでの学習から実際のバイオ分子へ
このアーキテクチャの上に、著者らは分子力のための「基盤モデル」であるLiTEN-FFを構築します。まず数百万の薬剤類似分子からなる大規模な量子化学データセットで学習させ、次にペプチド、溶媒和構造、ハロゲンや金属など多様な元素を含む、より高精度な小規模データで微調整します。この二段階学習により、幅広い化学領域のカバレッジと細部の精密さの両方を獲得します。高レベル量子結果と比較する標準ベンチマークでは、LiTENは小さな剛直分子から生物学的に関連する大規模系まで、多くの先行モデルと同等あるいはそれ以上の性能を示します。原子数が数百に達しても精度を維持し、多くの一般的な代替手法よりメモリ使用量が少なく、実行も高速です。
シミュレートされた化学での実用評価
静的なテストを越えて、チームは実際の研究ワークフローを模したタスクでもLiTEN-FFを評価します。薬剤に似た分子では、結果として得られる構造が要求の厳しい量子計算の結果とほぼ完全に一致するように形状を最適化できますが、速度は数千倍速くなります。分子動力学シミュレーションでは、結合長や角度の時間変動を再現し、量子ベースのシミュレーションを密接に追跡します。重要な結合まわりのねじれでエネルギーがどう変わるかという、構造選好性を捉えるために重要な予測でも優れています。液体水や水溶液中の短いペプチドでは、LiTEN-FFは実験やより高価な参照モデルと良く一致する構造的・熱力学的性質を生成し、大規模系では最大で十倍の高速化を実現します。
有用な形状探索の高速化
著者らはLiTEN-FFを中心に据えた実用的なコンフォーマー探索パイプラインも示します。高温ダイナミクス、冷却、簡易ジオメトリ精緻化を繰り返すことで、複雑な医薬分子に対して多様で低エネルギーな構造群を生成します。広く使われている量子ベースのワークフローと比較して、このAI駆動の手法はより多様なコンフォーマーを半分以下の時間で見つけます。さらにLiTEN-FFは多くの分子を並列処理してもコストが比例して増えないため、数千の候補を評価する大規模スクリーニングでは特に有力です。
今後の創薬・材料設計にとっての意味
要するに、LiTEN-FFは量子レベルの信頼性を日常利用にぐっと近づける新しい分子シミュレーションエンジンを提供します。角度やねじれの幾何情報を効率的なニューラルネットワークに直接符号化することで、高速だが近似的な力場と遅いが正確な量子計算とのギャップを埋めます。専門外の読者にとっての結論は、研究者がコンピュータ上でより現実的な分子“実験”を、現代の創薬や材料開発に見合った規模と速度で行えるようになる可能性が高いということです。広く採用されさらなる改良が進めば、この系統のモデルは合成前に新分子を提案・評価・精緻化する自動化パイプラインの中核になるかもしれません。
引用: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
キーワード: 分子シミュレーション, 機械学習力場, 創薬, バイオ分子モデリング, 量子化学