Clear Sky Science · fr
Un modèle de base extensible et d’exactitude quantique pour champs de force biomoléculaires via une attention quadrangulaire tensorisée linéairement
Pourquoi des « films » moléculaires plus rapides comptent
La découverte de médicaments et la conception de matériaux reposent de plus en plus sur des « films » informatiques montrant des molécules se tordre, se fléchir et réagir. Ces simulations peuvent révéler comment un médicament se loge dans la poche d’une protéine ou comment un nouveau matériau se comporte sous contrainte. Mais les outils actuels contraignent les scientifiques à choisir : des méthodes rapides qui sacrifient la précision, ou des calculs quantiques ultra-précis beaucoup trop lents pour des systèmes réalistes et complexes. Cet article présente LiTEN et son modèle de champ de force LiTEN-FF, une nouvelle approche par intelligence artificielle visant à fournir une précision de niveau quantique à des vitesses adaptées à la modélisation moléculaire courante.
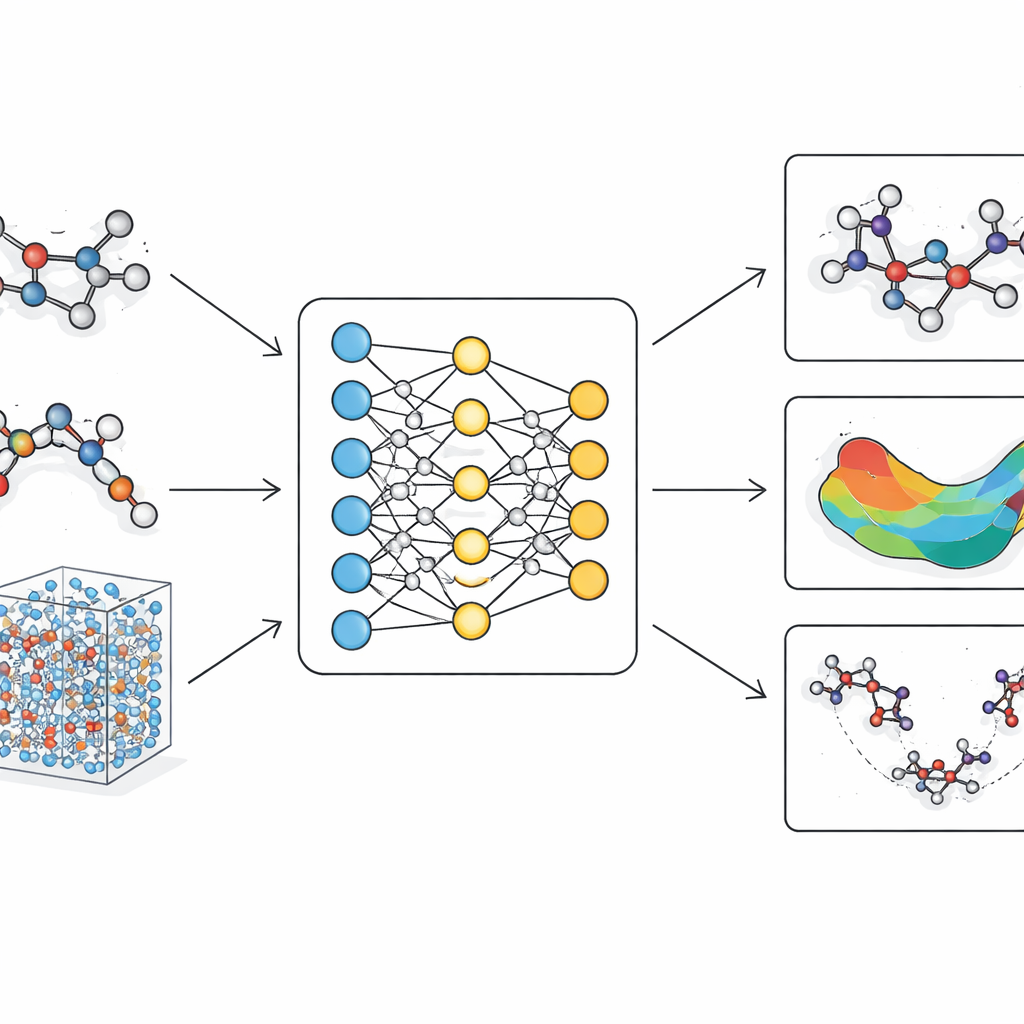
Le problème des modèles moléculaires actuels
Les simulations moléculaires traditionnelles se répartissent en deux catégories. Les champs de force classiques traitent les atomes comme des billes reliées par des ressorts aux paramètres fixes. Ils sont rapides et capables de gérer de grandes protéines ou de longues échelles de temps, mais peinent à rendre des changements de forme subtils, des réarrangements de liaisons et des voies réactionnelles importants en chimie réelle. Les méthodes quantiques, en revanche, décrivent explicitement les électrons et peuvent capturer avec précision la rupture et la formation de liaisons. Toutefois, elles sont si coûteuses en calcul qu’elles se limitent généralement à de petites molécules ou à des simulations très courtes. Ces dernières années, des approches d’apprentissage automatique ont émergé comme voie intermédiaire, apprenant à imiter les calculs quantiques. Beaucoup de ces modèles manquent toutefois de rigueur physique nécessaire à la fiabilité ou deviennent trop lents lorsqu’on les étend à de grandes biomolécules réalistes.
Une nouvelle manière d’apprendre à l’IA la géométrie moléculaire
LiTEN relève ce défi en repensant la façon dont un réseau neuronal « ressent » la géométrie moléculaire. Plutôt que de ne considérer que des distances par paires simples entre atomes, LiTEN intègre des informations sur des motifs à trois et quatre atomes qui contrôlent les angles et les torsions d’une molécule. Fait crucial, cela respecte les symétries physiques de base : si l’on fait tourner ou déplacer une molécule dans l’espace, son énergie prédite reste identique et les forces prédites tournent de manière appropriée. L’innovation clé, appelée attention quadrangulaire tensorisée, permet au modèle de capturer des interactions complexes de flexion et de torsion en utilisant des opérations vectorielles efficaces plutôt que des mathématiques lourdes et spécialisées. Cela rend le calcul évolutif, de sorte que les effets multi-corps qui ralentissent habituellement les modèles avancés peuvent être traités avec un coût qui croît seulement linéairement avec la taille du système.
De l’entraînement sur données quantiques aux biomolécules réelles
À partir de cette architecture, les auteurs construisent LiTEN-FF, un « modèle de base » pour les forces moléculaires. Ils l’entraînent d’abord sur un jeu de données massif de chimie quantique comprenant des millions de molécules de type médicament, puis le raffinent sur un ensemble plus petit mais de plus haute précision incluant des peptides, des structures solvatées et des éléments divers tels que halogènes et métaux. Cet entraînement en deux étapes permet au modèle d’acquérir à la fois une large couverture chimique et des détails fins. Dans des tests de référence standard comparant énergies et forces prédites à des résultats quantiques de haut niveau, LiTEN égale ou dépasse les modèles leaders, tant pour des petites molécules rigides que pour des systèmes biologiquement pertinents bien plus grands. Il conserve son exactitude même lorsque le nombre d’atomes atteint les centaines, tout en consommant moins de mémoire et en s’exécutant plus rapidement que de nombreuses alternatives populaires.
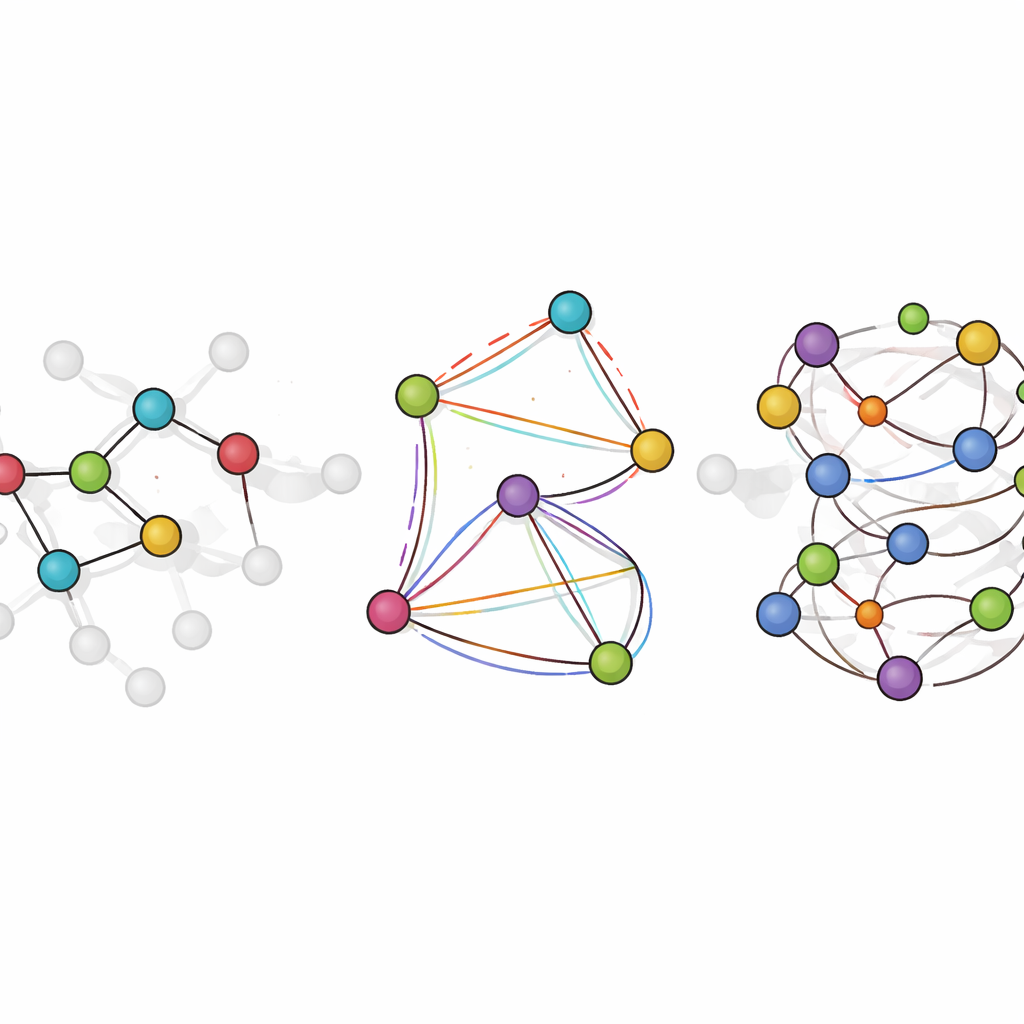
Mettre le modèle en pratique en chimie simulée
Au-delà des tests statiques, l’équipe évalue LiTEN-FF sur des tâches qui reflètent des flux de travail de recherche réels. Pour des molécules de type médicament, il peut optimiser les conformations de sorte que les structures résultantes correspondent presque parfaitement à celles obtenues par des calculs quantiques exigeants, mais des milliers de fois plus vite. Lors de simulations de dynamique moléculaire, il reproduit la façon dont les longueurs de liaison et les angles fluctuent dans le temps, en suivant de près les simulations basées sur la mécanique quantique. Il excelle également à prédire comment l’énergie varie lorsqu’une molécule se tord autour d’une liaison clé, un ingrédient crucial pour saisir les préférences conformationnelles en conception de médicaments. Dans l’eau liquide et pour de courts peptides dissous, LiTEN-FF génère des propriétés structurelles et thermodynamiques en bon accord avec les expériences et les modèles de référence plus coûteux, tout en offrant jusqu’à un gain de vitesse d’un facteur dix sur de grands systèmes.
Accélérer la recherche de formes utiles
Les auteurs démontrent aussi une chaîne pratique de recherche de conformères construite autour de LiTEN-FF. En exécutant des cycles répétés de dynamique à haute température, de refroidissement et de raffinement géométrique rapide, le modèle génère des ensembles riches de formes distinctes et à basse énergie pour des molécules médicamenteuses complexes. Comparée à une chaîne de traitement quantique largement utilisée, cette approche pilotée par l’IA trouve des conformères plus diversifiés en moins de la moitié du temps. De plus, parce que LiTEN-FF peut traiter de nombreuses molécules en parallèle sans augmentation proportionnelle du coût, il devient particulièrement puissant pour des campagnes de criblage à grande échelle évaluant des milliers de candidats.
Ce que cela signifie pour la conception future de médicaments et de matériaux
En substance, LiTEN-FF propose un nouveau moteur pour la simulation moléculaire qui rapproche fortement la fiabilité de niveau quantique de l’usage quotidien. En encodant la géométrie des angles et des torsions directement dans un réseau neuronal efficace, il réduit l’écart entre des champs de force rapides mais approximatifs et des calculs quantiques lents mais précis. Pour les non-spécialistes, la conclusion est que les chercheurs pourront bientôt exécuter des « expériences » moléculaires informatiques beaucoup plus réalistes, à des échelles et des vitesses compatibles avec la découverte moderne de médicaments et le développement de matériaux. Si ces modèles sont largement adoptés et affinés, ils pourraient devenir des composants centraux de pipelines automatisés proposant, testant et raffinant de nouvelles molécules bien avant leur synthèse en laboratoire.
Citation: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Mots-clés: simulation moléculaire, champs de force appris par machine, découverte de médicaments, modélisation biomoléculaire, chimie quantique