Clear Sky Science · zh
一个端到端、可泛化的深度学习框架,用于全面分析转录调控
在不做每项实验的情况下“读懂”DNA
现代生物学通常需要数十项昂贵的实验来绘制每种细胞类型中基因如何被调控的图谱。本研究展示了如何通过将测序数据与人工智能巧妙结合,用一个方法替代许多实验,提供一种更快、更便宜的方式来读取基因组的控制系统。
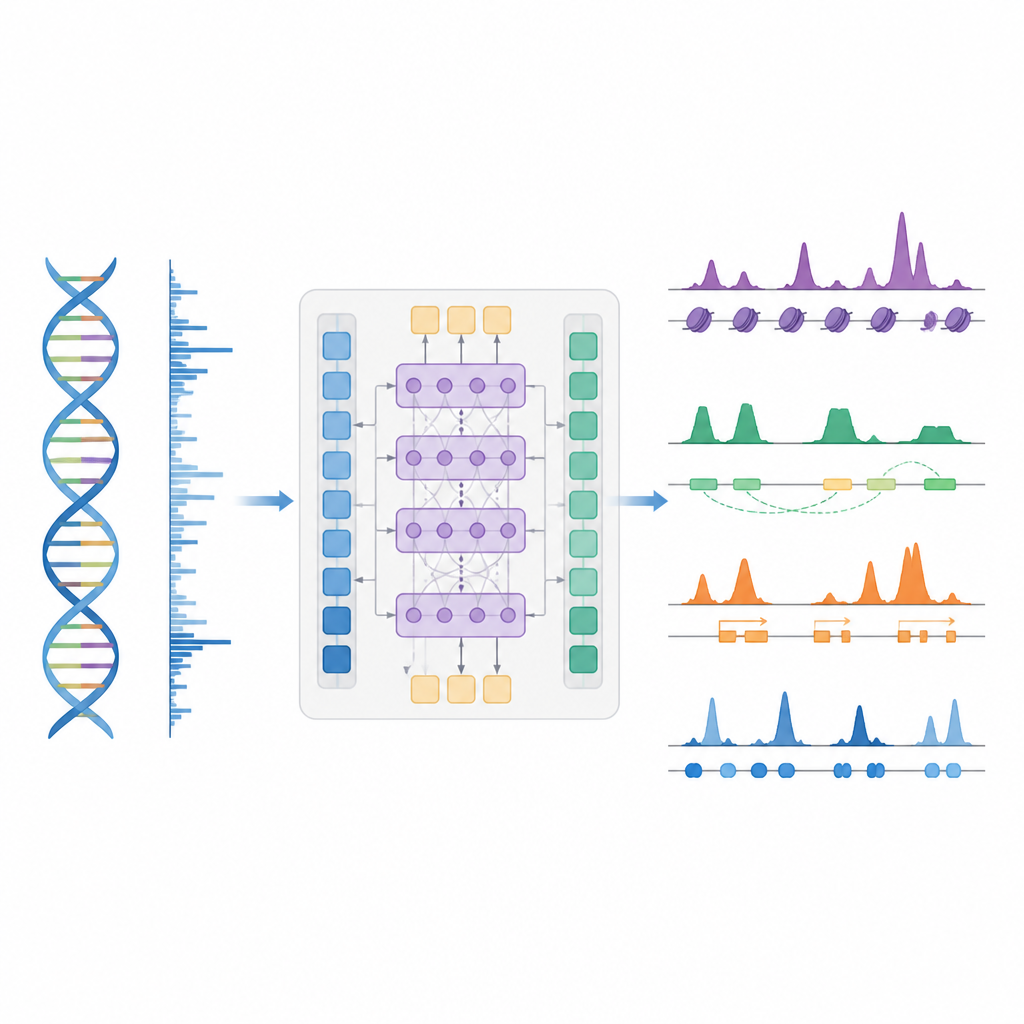
更聪明的捷径来绘制基因调控图
作者提出了 BioSeq2Seq,一种旨在从基因组推断多种调控信息的深度学习框架。与其为每种化学修饰或蛋白质重复独立实验,BioSeq2Seq 从两类主要输入中学习。一类是 DNA 序列本身,它在几乎所有细胞中都相同;另一类是来自 run-on 测序检测的数据显示 RNA 聚合酶在 DNA 上活动的位置及方向。这类测定捕获了特定细胞类型中基因组哪些部分正在被使用的即时快照。通过结合这两种信息源,模型能够预测生物学家通常用不同实验测量的广泛特征。
模型如何在基因组中识别模式
BioSeq2Seq 依赖于 transformer 架构——一种最初用于语言模型的神经网络类型。在这里,“语言”是 DNA 的碱基序列加上沿染色体分布的转录信号模式。模型首先将 DNA 和 run-on 信号都转换为数值特征,然后使用注意力层来连接跨越十万多个碱基的远距离位点。这种远视角很重要,因为如增强子等调控元件可以在很远处作用于它们所调控的基因。基于学习到的这些模式,模型在基因组上输出精细间距的预测,例如组蛋白上的激活或沉默标记可能出现的位置、转录何处起始或终止以及特定蛋白质倾向于结合的位置。
在多种细胞、组织与物种上的测试
研究人员主要在一种人类血液癌细胞系的数据上训练 BioSeq2Seq,然后在许多其他情境中对其进行检验。这些情境包括若干人类细胞类型、小鼠和马的肝脏及果蝇的卵巢。在十种组蛋白标记中,模型的预测与实验测量高度吻合,尤其是与活性基因相关的标记。它在基因起始位点周围以及启动子和增强子区域也表现良好——这些区域的基因调控尤为强烈。与早期使用更简单统计模型或更少数据类型的工具相比,BioSeq2Seq 在组蛋白标记上的平均准确度提高了超过 14%,且速度更快,因为它能够一次性预测所有标记,而不是逐个预测。
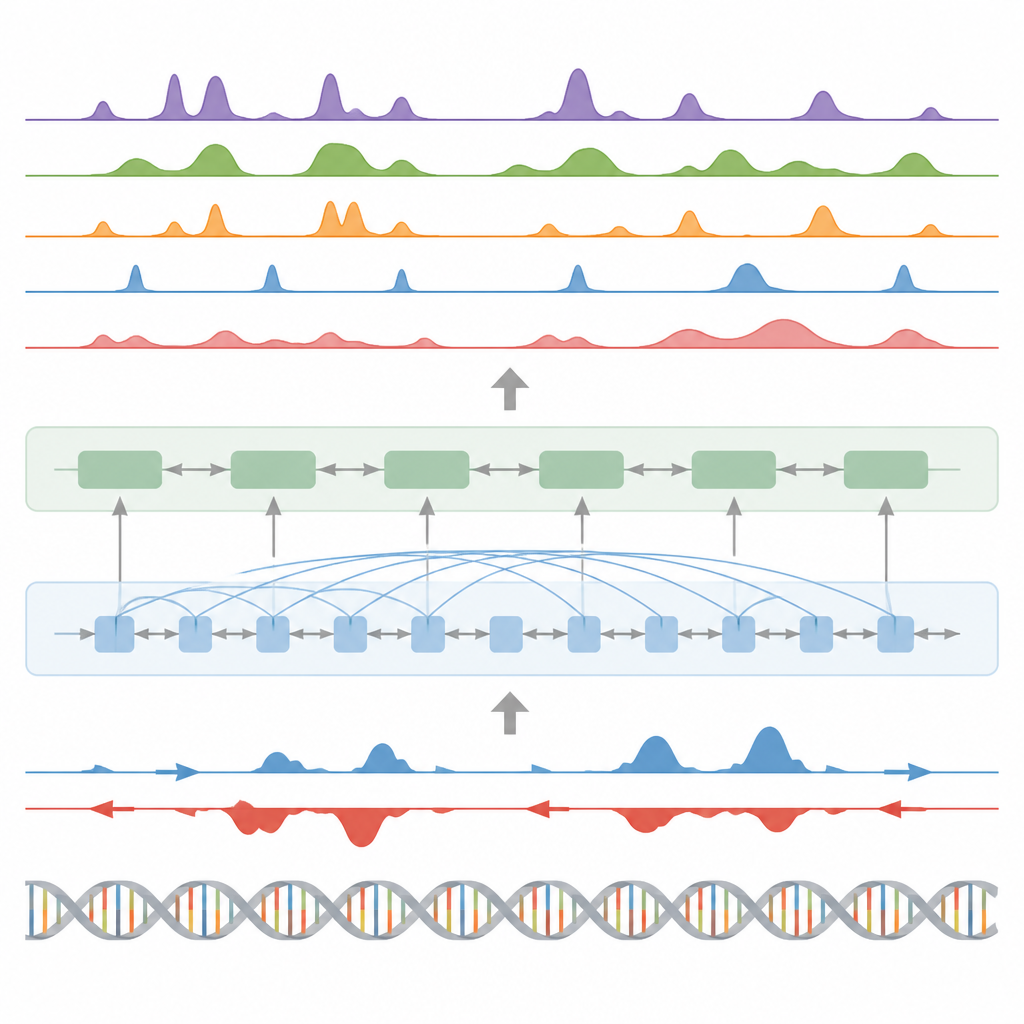
识别关键开关、基因活性和蛋白“足迹”
除了组蛋白标记外,模型还在三项主要任务上进行了测试。首先,它通过将连续信号预测转换为峰值并使用自定义统计峰值检测器,识别了功能元件,如转录起始区、绝缘子、poly(A) 位点和完整的基因体。对于起始区和基因体,模型在准确率和召回率上都取得了很高的得分,并且优于一种广泛使用的活性调控位点识别方法。第二,BioSeq2Seq 预测了完整的基因表达谱,而不仅仅是高/低分类,基于其输出建立的一个简单分类器击败了若干依赖更多实验输入的领先模型。第三,使用相同框架,作者训练系统预测九十种不同转录因子的结合位点,性能可比拟于一种使用开放染色质数据的顶级方法,且在最难预测的因子上有所改进,同时仅使用单一共享模型。
这对基因组研究意味着什么
通过学习 DNA 序列与单一转录测定如何对应于多层次的基因调控,BioSeq2Seq 提供了运行数十项独立实验的实用替代方案。它允许研究者在仅有 run-on 数据和参考基因组的新细胞类型、组织乃至物种中推断组蛋白标记、调控元件、基因活性和蛋白结合情况。对非专业读者而言,关键信息是:一项精心选择的实验,结合强大的学习系统,如今可替代一整套昂贵的检测工具,使大规模的基因调控研究对更多实验室和生物学问题可及。
引用: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
关键词: 基因调控, 深度学习, 基因组注释, 转录, 表观基因组学