Clear Sky Science · de
Ein durchgängiges, generalisierbares Deep‑Learning‑Framework zur umfassenden Analyse der Transkriptionsregulation
DNA lesen, ohne jede Labortest‑Reihe durchzuführen
Die moderne Biologie benötigt häufig Dutzende kostspieliger Laborexperimente, um zu erfassen, wie Gene in jedem Zelltyp reguliert werden. Diese Studie zeigt, wie eine einzige clevere Kombination aus Sequenzierungsdaten und künstlicher Intelligenz viele dieser Tests ersetzen kann und so eine schnellere und günstigere Methode bietet, das Kontrollsystem des Genoms zu entschlüsseln.
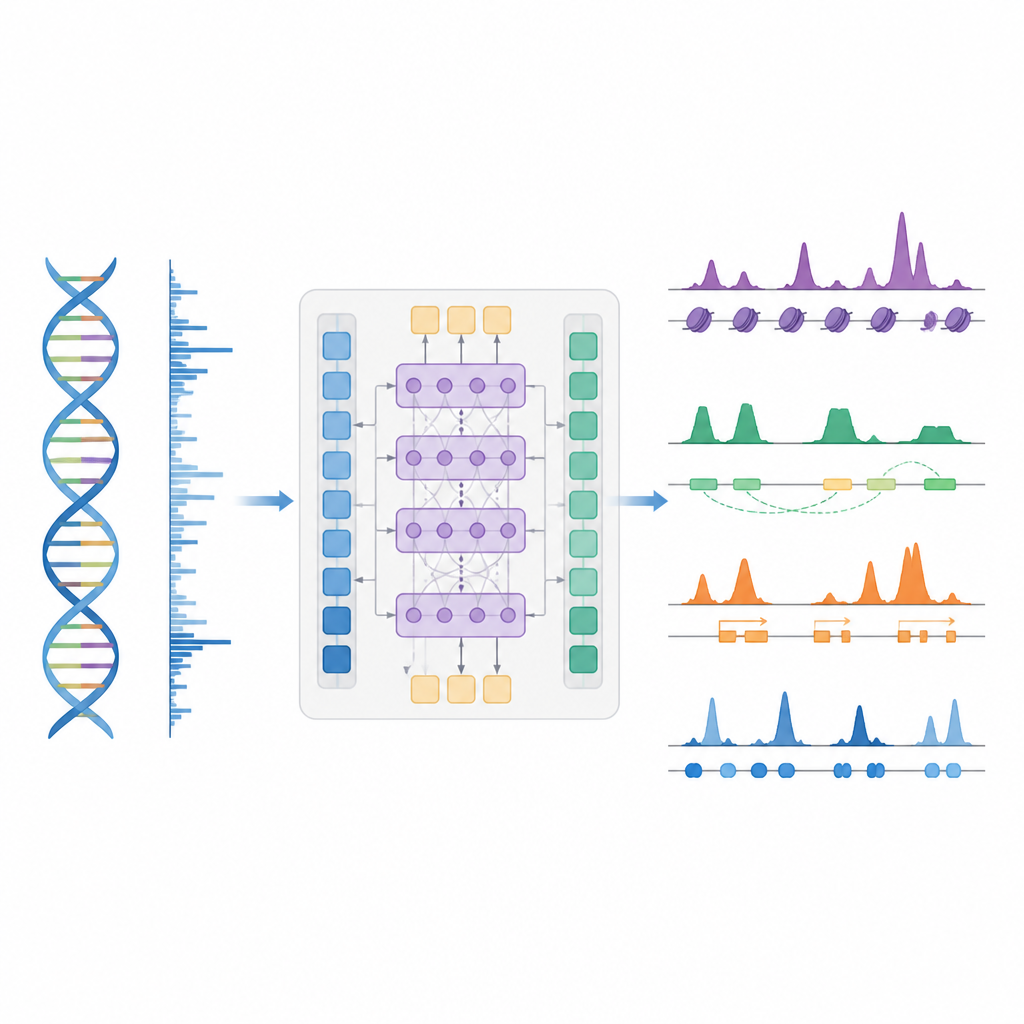
Eine intelligentere Abkürzung zur Kartierung der Genkontrolle
Die Autor*innen stellen BioSeq2Seq vor, ein Deep‑Learning‑Framework, das entwickelt wurde, um viele Arten regulatorischer Informationen aus dem Genom abzuleiten. Anstatt für jedes chemische Mark oder Protein separate Experimente zu wiederholen, lernt BioSeq2Seq aus zwei Hauptinputs. Der eine ist die DNA‑Sequenz selbst, die in praktisch allen Zellen gleich ist. Der andere sind Daten aus einem Run‑On‑Sequencing‑Assay, der anzeigt, wo die RNA‑Polymerase aktiv entlang der DNA voranschreitet und in welche Richtung. Dieser Assay liefert eine Momentaufnahme, welche Bereiche des Genoms in einem bestimmten Zelltyp genutzt werden. Durch die Kombination dieser beiden Quellen kann das Modell eine breite Palette von Merkmalen vorhersagen, die Biolog*innen üblicherweise mit getrennten Experimenten messen.
Wie das Modell Muster im Genom erkennt
BioSeq2Seq basiert auf einer Transformer‑Architektur, einer Art neuronalen Netzes, das ursprünglich in Sprachmodellen verwendet wurde. Hier ist die „Sprache“ die Abfolge von Basen in der DNA plus das Muster der Transkriptionssignale entlang des Chromosoms. Das Modell wandelt zuerst sowohl DNA als auch Run‑On‑Signale in numerische Merkmale um und verwendet dann Attention‑Schichten, die entfernte Stellen über mehr als 100.000 DNA‑Basen hinweg verbinden können. Dieser weite Blick ist wichtig, weil Kontroll‑Elemente wie Enhancer weit von den Genen wirken können, die sie regulieren. Aus diesen gelernten Mustern erzeugt das Modell fein gerasterte Vorhersagen entlang des Genoms, etwa wo aktivierende oder stillende Histonmodifikationen zu erwarten sind, wo die Transkription beginnt oder stoppt und wo sich bestimmte Proteine bevorzugt binden.
Testen über viele Zellen, Gewebe und Arten hinweg
Die Forschenden trainierten BioSeq2Seq hauptsächlich mit Daten aus einer menschlichen Blutkrebs‑Zelllinie und prüften es dann in vielen anderen Kontexten. Dazu gehörten mehrere menschliche Zelltypen, Lebergewebe von Maus und Pferd sowie Eierstöcke der Fruchtfliege. Für zehn Typen von Histonmarken stimmten die Modellvorhersagen eng mit experimentellen Messungen überein, besonders für Markierungen, die mit aktiven Genen verbunden sind. Es lieferte auch gute Ergebnisse in Regionen um Transkriptionsstartstellen sowie in Promotoren und Enhancern, wo Genkontrolle am intensivsten ist. Im Vergleich zu früheren Werkzeugen, die einfachere statistische Modelle oder weniger Datentypen nutzten, verbesserte BioSeq2Seq die Genauigkeit für Histonmarken im Mittel um mehr als 14 Prozent und tat dies deutlich schneller, indem es alle Markierungen gleichzeitig statt einzeln vorhersagte.
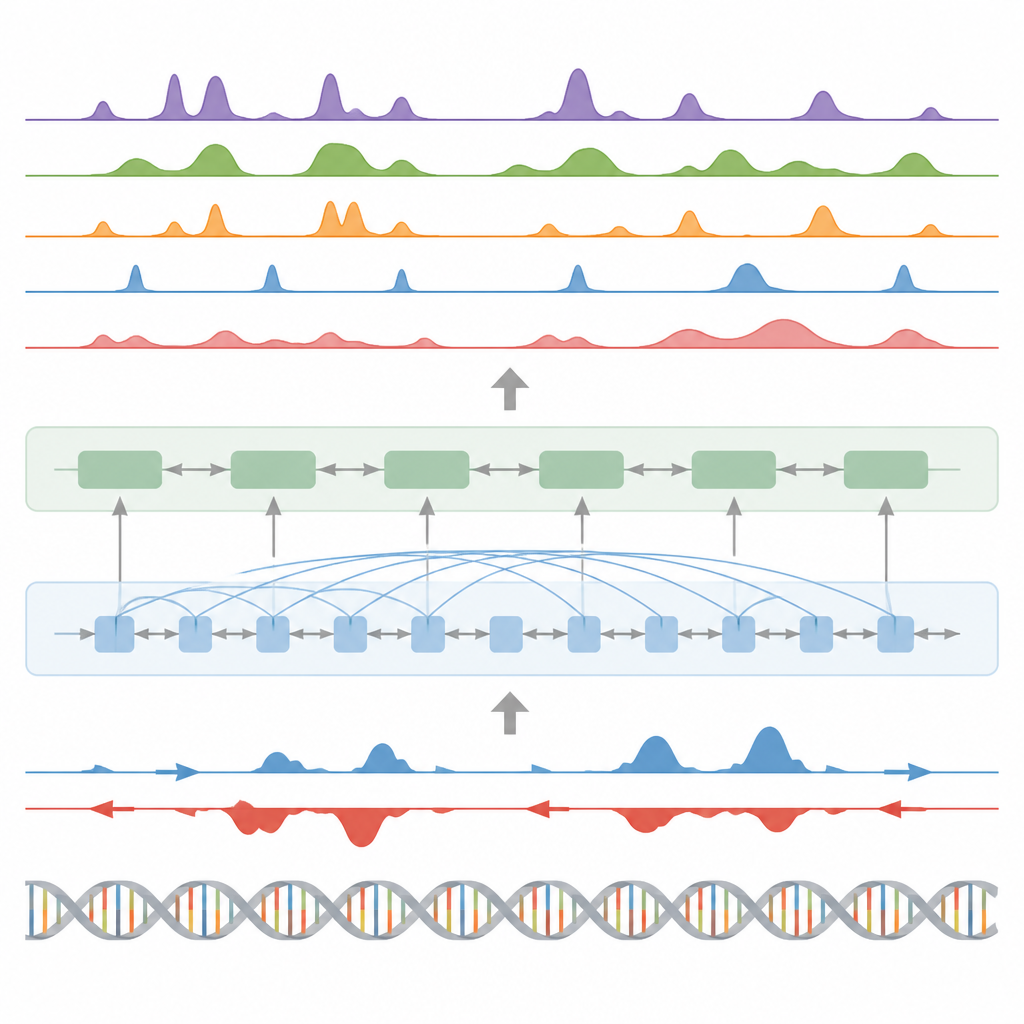
Schlüsselschalter, Genaktivität und Proteinfußabdrücke finden
Über Histonmarken hinaus wurde das Modell an drei weiteren zentralen Aufgaben getestet. Erstens identifizierte es funktionelle Elemente wie Transkriptionsstartregionen, Insulatoren, Poly(A)‑Stellen und ganze Genkörper, indem es seine kontinuierlichen Signalvorhersagen mit einem eigenen statistischen Peak‑Caller in Peaks umwandelte. Für Startregionen und Genkörper erzielte es hohe Werte für Genauigkeit und Wiederauffindungsrate und übertraf eine weit verbreitete Methode zur Erkennung aktiver regulatorischer Stellen. Zweitens sagte BioSeq2Seq vollständige Genexpressionsprofile vorher, nicht nur hoch versus niedrig; ein einfacher Klassifikator auf Basis dieser Ausgaben schlug mehrere führende Modelle, die weitaus mehr experimentelle Eingaben benötigen. Drittens trainierten die Autor*innen mit demselben Framework Vorhersagen für Bindungsstellen von neunzig verschiedenen Transkriptionsfaktoren und erreichten eine Leistung, die mit einer Spitzenmethode auf Basis offener Chromatin‑Daten vergleichbar ist und bei den schwierigsten Faktoren sogar Verbesserungen zeigte, während nur ein einziges gemeinsames Modell verwendet wurde.
Was das für die Genomforschung bedeutet
Indem BioSeq2Seq erlernt, wie DNA‑Sequenz und ein einziger Transkriptionsassay mit vielen Ebenen der Genkontrolle zusammenhängen, bietet es eine praktische Alternative zum Durchführen dutzender einzelner Experimente. Forschende können damit Histonmarken, regulatorische Elemente, Genaktivität und Proteingebundenheit in neuen Zelltypen, Geweben und sogar Arten ableiten, in denen nur Run‑On‑Daten und ein Referenzgenom verfügbar sind. Für eine nichtfachliche Leserschaft lautet die Kernaussage: Ein sorgfältig gewähltes Experiment kombiniert mit einem leistungsfähigen Lernsystem kann nun eine ganze Werkzeugkiste teurer Tests ersetzen und groß angelegte Studien zur Genregulation für viele weitere Labore und Fragestellungen zugänglich machen.
Zitation: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
Schlüsselwörter: Genregulation, Deep Learning, Genomanotation, Transkription, Epigenomik