Clear Sky Science · es
Un marco de aprendizaje profundo generalizable de extremo a extremo para analizar de forma integral la regulación transcripcional
Leer el ADN sin realizar cada prueba de laboratorio
La biología moderna a menudo requiere docenas de experimentos de laboratorio costosos para mapear cómo se controlan nuestros genes en cada tipo celular. Este estudio muestra cómo una sola combinación ingeniosa de datos de secuenciación e inteligencia artificial puede sustituir a muchas de esas pruebas, ofreciendo una forma más rápida y barata de leer el sistema de control del genoma.
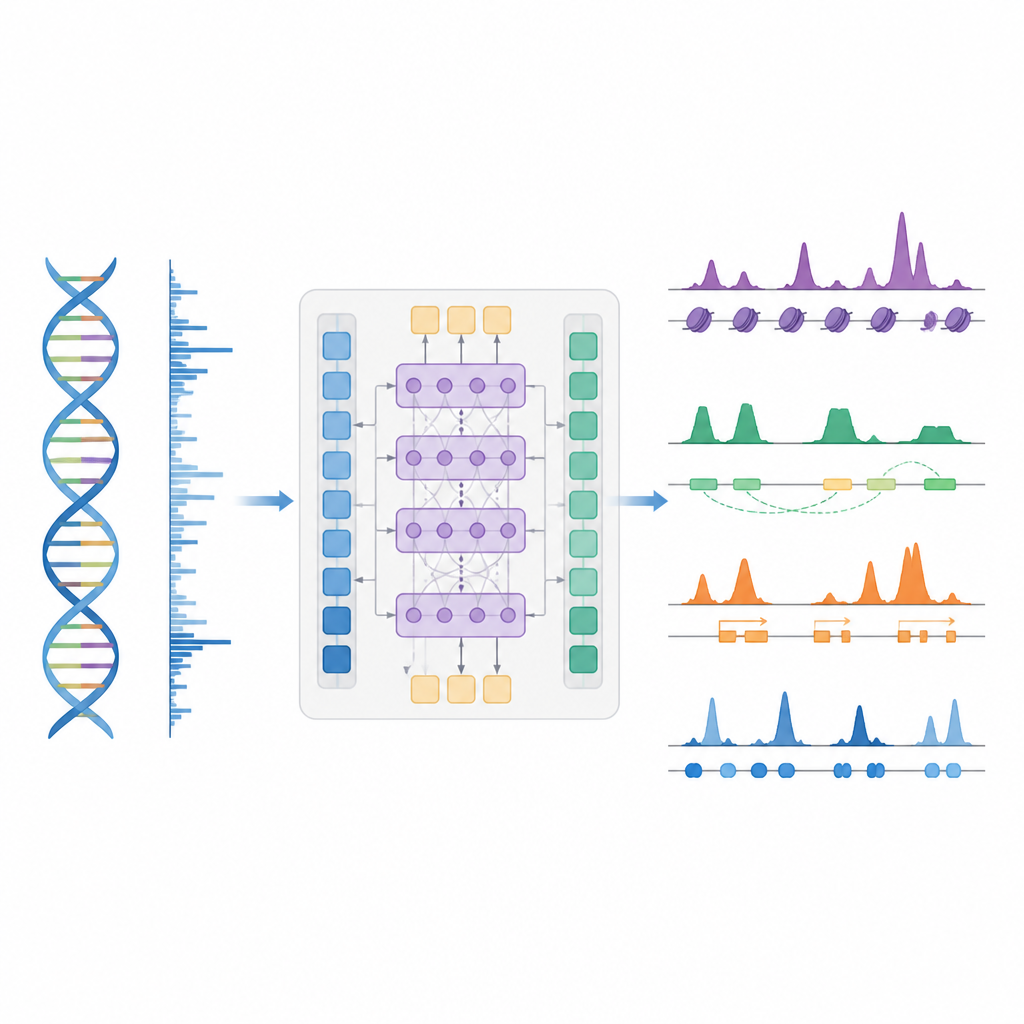
Un atajo más inteligente para mapear el control génico
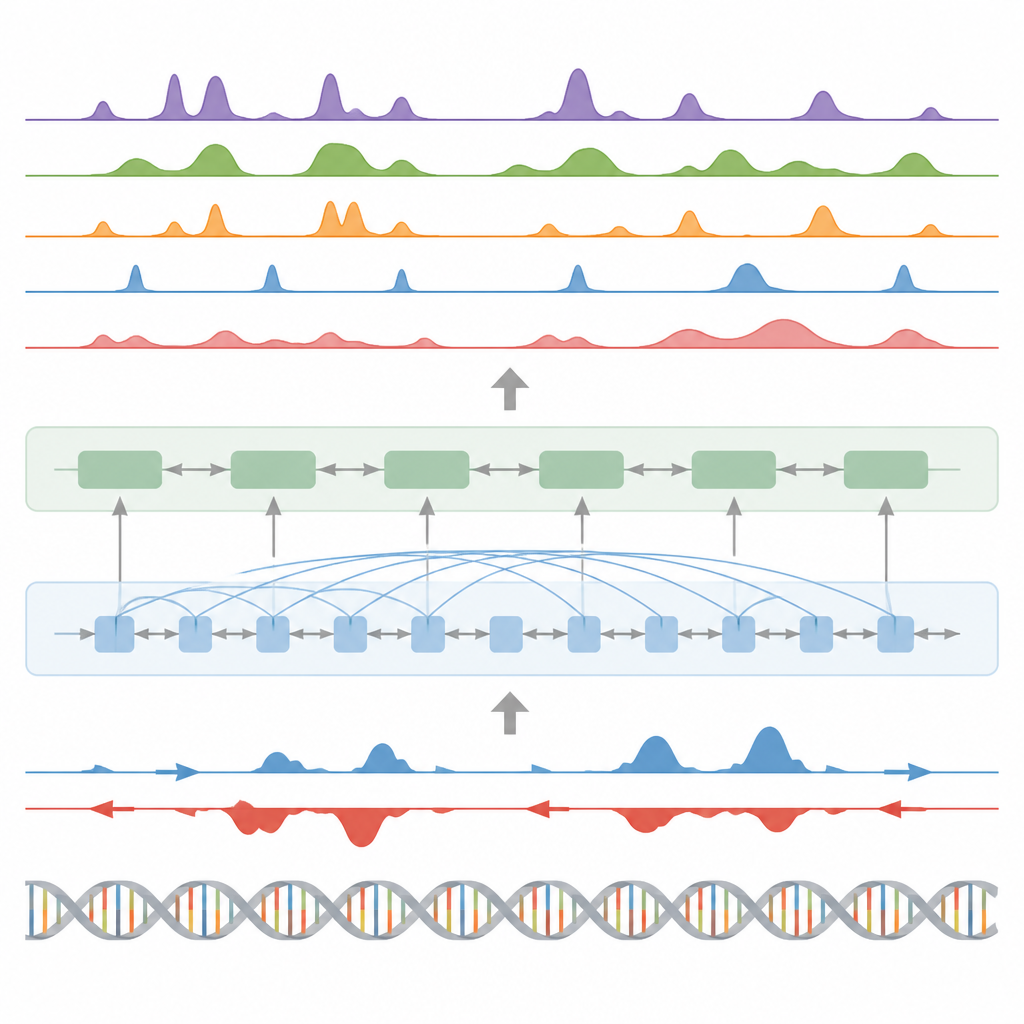
Los autores presentan BioSeq2Seq, un marco de aprendizaje profundo diseñado para inferir muchos tipos de información regulatoria a partir del genoma. En lugar de repetir experimentos separados para cada etiqueta química o proteína, BioSeq2Seq aprende a partir de dos entradas principales. Una es la propia secuencia de ADN, que es la misma en casi todas las células. La otra son los datos de un ensayo de secuenciación run-on que informa dónde la ARN polimerasa se está moviendo activamente a lo largo del ADN y en qué dirección. Este ensayo captura una instantánea en vivo de qué partes del genoma se están utilizando en un tipo celular específico. Al combinar estas dos fuentes, el modelo puede predecir una amplia gama de características que los biólogos normalmente miden con experimentos separados.
Cómo el modelo detecta patrones en el genoma
BioSeq2Seq se basa en una arquitectura transformer, un tipo de red neuronal usado originalmente en modelos de lenguaje. Aquí, el “lenguaje” es la secuencia de bases del ADN más el patrón de señales de transcripción a lo largo del cromosoma. El modelo primero convierte tanto el ADN como las señales run-on en características numéricas y luego utiliza capas de atención que pueden conectar sitios distantes a través de más de 100.000 letras de ADN. Esta visión a largo alcance es importante porque elementos de control como los potenciadores pueden actuar lejos de los genes que regulan. A partir de estos patrones aprendidos, el modelo genera predicciones finamente espaciadas a lo largo del genoma, como dónde deberían aparecer marcas de histonas que activan o silencian, dónde comienza o termina la transcripción y dónde tienden a unirse proteínas específicas.
Pruebas en muchas células, tejidos y especies
Los investigadores entrenaron BioSeq2Seq principalmente con datos de una línea celular humana de cáncer sanguíneo y luego lo evaluaron en muchos otros contextos. Estos incluyeron varios tipos celulares humanos, hígado de ratón y caballo, y ovario de mosca de la fruta. En diez tipos de marcas de histonas, las predicciones del modelo coincidieron estrechamente con las mediciones experimentales, especialmente para las marcas vinculadas a genes activos. También funcionó bien en regiones alrededor de los sitios de inicio génico y dentro de promotores y potenciadores, donde el control génico es más intenso. En comparación con herramientas anteriores que usaban modelos estadísticos más simples o menos tipos de datos, BioSeq2Seq mejoró la precisión para marcas de histonas en más de un 14 % en promedio y lo hizo mucho más rápido, prediciendo todas las marcas a la vez en lugar de una por una.
Encontrar interruptores clave, actividad génica y huellas de proteínas
Más allá de las marcas de histonas, el modelo se probó en otras tres tareas principales. Primero, identificó elementos funcionales como regiones de inicio de transcripción, aisladores, sitios de poli(A) y cuerpos génicos completos convirtiendo sus predicciones de señal continua en picos mediante un detector de picos estadístico personalizado. Para regiones de inicio y cuerpos génicos alcanzó puntuaciones altas tanto en precisión como en recuperación y superó a un método ampliamente usado para encontrar sitios regulatorios activos. Segundo, BioSeq2Seq predijo perfiles completos de expresión génica, no solo alto frente a bajo, y luego un clasificador simple construido sobre sus salidas superó a varios modelos líderes que dependen de muchos más insumos experimentales. Tercero, usando el mismo marco, los autores entrenaron el sistema para predecir sitios de unión de noventa factores de transcripción diferentes, alcanzando un rendimiento similar al de un método de primera línea que usa datos de cromatina abierta e incluso mejorando en los factores más difíciles mientras empleaba un único modelo compartido.
Qué significa esto para el estudio de genomas
Al aprender cómo la secuencia de ADN y un único ensayo de transcripción se relacionan con muchas capas de control génico, BioSeq2Seq ofrece una alternativa práctica a realizar docenas de experimentos separados. Permite a los investigadores inferir marcas de histonas, elementos regulatorios, actividad génica y unión de proteínas en nuevos tipos celulares, tejidos e incluso especies donde solo están disponibles datos run-on y un genoma de referencia. Para un lector no especialista, el mensaje clave es que un experimento cuidadosamente elegido, combinado con un sistema de aprendizaje potente, puede ahora sustituir a todo un conjunto de pruebas costosas, acercando los estudios a gran escala de la regulación génica a muchos más laboratorios y preguntas biológicas.
Cita: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
Palabras clave: regulación génica, aprendizaje profundo, anotación del genoma, transcripción, epigenómica