Clear Sky Science · nl
Een end-to-end generaliseerbaar deep learning-kader voor een alomvattende analyse van transcriptieregulatie
DNA lezen zonder elk laboratoriumtest uit te voeren
De moderne biologie heeft vaak tientallen kostbare laboratoriumexperimenten nodig om in kaart te brengen hoe onze genen in elk celtype worden geregeld. Deze studie toont aan hoe een enkele slimme combinatie van sequencinggegevens en kunstmatige intelligentie kan optreden als vervanging voor veel van die tests, en zo een snellere en goedkopere manier biedt om het regelsysteem van het genoom te doorgronden.
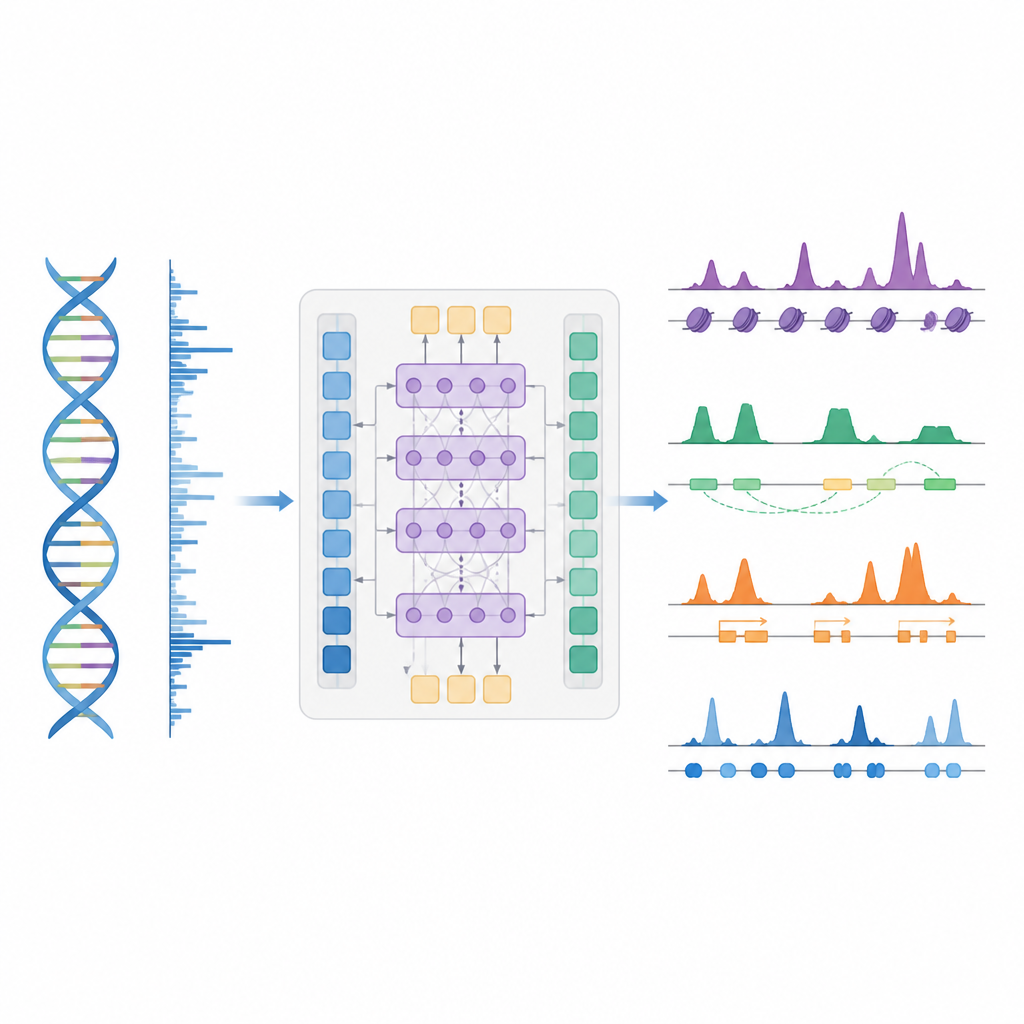
Een slimmer kortere weg om genregulatie te kaartbrengen
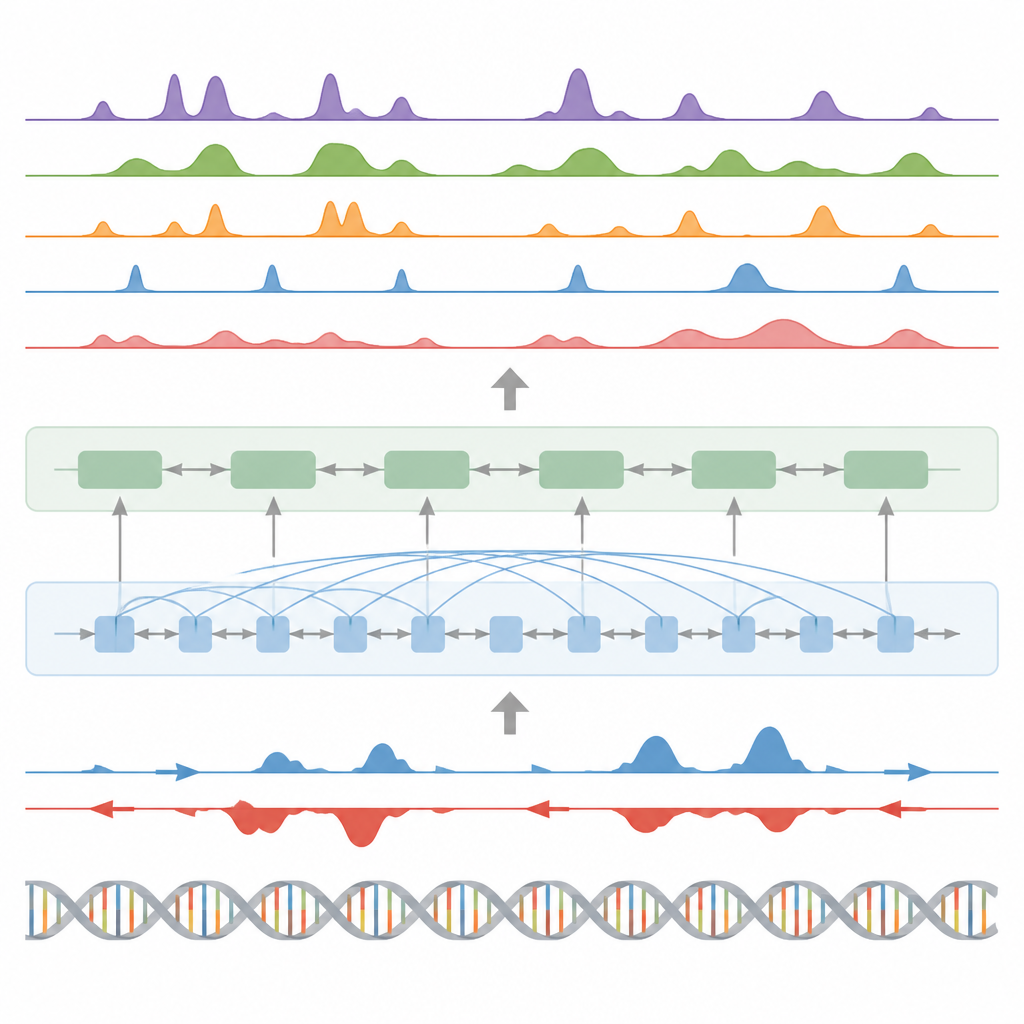
De auteurs introduceren BioSeq2Seq, een deep learning-kader ontworpen om veel soorten regulerende informatie af te leiden uit het genoom. In plaats van voor elk chemisch label of eiwit afzonderlijke experimenten te herhalen, leert BioSeq2Seq uit twee hoofdinputs. De ene is de DNA-sequentie zelf, die in vrijwel alle cellen hetzelfde is. De andere is data van een run-on sequencingassay die rapporteert waar RNA-polymerase actief langs het DNA beweegt en in welke richting. Deze assay legt een momentopname vast van welke delen van het genoom in een specifiek celtype worden gebruikt. Door deze twee bronnen te combineren, kan het model een breed scala aan kenmerken voorspellen die biologen normaal gesproken met afzonderlijke experimenten meten.
Hoe het model patronen in het genoom ziet
BioSeq2Seq vertrouwt op een transformer-architectuur, een type neuraal netwerk dat oorspronkelijk is ontwikkeld voor taalmodellen. Hier is de "taal" de volgorde van basen in DNA plus het patroon van transcriptiesignalen langs het chromosoom. Het model zet eerst zowel DNA als run-on-signalen om in numerieke kenmerken en gebruikt vervolgens attentielaaglagen die verre locaties kunnen verbinden over meer dan 100.000 DNA-letters. Dit langdurige zicht is belangrijk omdat controle-elementen zoals enhancers ver van de genen waarop ze inwerken kunnen liggen. Vanuit deze aangeleerde patronen genereert het model fijnmazige voorspellingen langs het genoom, zoals waar activerende of stillegende merkers op histoneiwitten zouden moeten verschijnen, waar transcriptie begint of stopt, en waar specifieke eiwitten neigen te binden.
Testen over vele cellen, weefsels en soorten
De onderzoekers trainden BioSeq2Seq voornamelijk op data van één menselijke bloedkankercellijn, en daagden het model vervolgens uit in vele andere contexten. Daartoe behoorden meerdere menselijke celtypen, lever van muis en paard, en eierstok van de fruitvlieg. Over tien typen histonmerken kwamen de voorspellingen van het model goed overeen met experimentele metingen, vooral voor merkers die aan actieve genen zijn gekoppeld. Het behaalde ook goede resultaten in regio's rond genstartplaatsen en binnen promotors en enhancers, waar genregulatie het meest intens is. Vergeleken met eerdere tools die eenvoudigere statistische modellen of minder datatypen gebruikten, verbeterde BioSeq2Seq de nauwkeurigheid voor histonmerken met gemiddeld meer dan 14 procent en deed dit veel sneller doordat het alle merkers tegelijk voorspelt in plaats van één voor één.
Belangrijke schakelaars, genactiviteit en eiwitvoetafdrukken vinden
Naast histonmerken werd het model getest op drie andere grote taken. Ten eerste identificeerde het functionele elementen zoals transcriptiestartgebieden, insulators, poly(A)-sites en volledige genlichamen door zijn continue signaalsvoorspellingen om te zetten in pieken met een aangepaste statistische piekdetector. Voor startregio's en genlichamen behaalde het hoge scores voor zowel nauwkeurigheid als recall en overtrof het een veel gebruikte methode voor het vinden van actieve regulerende sites. Ten tweede voorspelde BioSeq2Seq volledige genexpressieprofielen, niet alleen hoog versus laag, en een eenvoudige classifier gebouwd op zijn output versloeg vervolgens meerdere toonaangevende modellen die veel meer experimentele inputs gebruiken. Ten derde gebruikten de auteurs hetzelfde kader om het systeem te trainen om bindingssites voor negentig verschillende transcriptiefactoren te voorspellen, met prestaties die vergelijkbaar zijn met een topmethode die open-chromatine-data gebruikt en zelfs verbetering liet zien voor de moeilijkste factoren, terwijl er slechts één gedeeld model werd gebruikt.
Wat dit betekent voor het bestuderen van genomen
Door te leren hoe DNA-sequentie en één enkele transcriptieassay samenhangen met vele lagen van genregulatie, biedt BioSeq2Seq een praktische alternatieve route voor het uitvoeren van tientallen afzonderlijke experimenten. Het stelt onderzoekers in staat histonmerken, regulerende elementen, genactiviteit en eiwitbinding af te leiden in nieuwe celtypen, weefsels en zelfs soorten waar alleen run-on-data en een referentiegenoom beschikbaar zijn. Voor de niet-specialistische lezer is de kernboodschap dat één zorgvuldig gekozen experiment, gecombineerd met een krachtig leersysteem, nu kan dienen als vervanging voor een hele gereedschapskist aan kostbare tests, waardoor grootschalige studies naar genregulatie binnen het bereik van veel meer laboratoria en biologische vragen komen.
Bronvermelding: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
Trefwoorden: genregulatie, deep learning, genoomannotatie, transcriptie, epigenomica