Clear Sky Science · pl
Kompleksowe, uogólnialne rozwiązanie głębokiego uczenia do całościowej analizy regulacji transkrypcyjnej
Odczytywanie DNA bez wykonywania wszystkich badań laboratoryjnych
Współczesna biologia często wymaga dziesiątek kosztownych eksperymentów laboratoryjnych, aby zmapować, jak geny są kontrolowane w każdym typie komórek. To badanie pokazuje, że pojedyncze sprytne połączenie danych sekwencyjnych i sztucznej inteligencji może zastąpić wiele z tych testów, oferując szybszy i tańszy sposób „odczytu” systemu kontroli genomu.
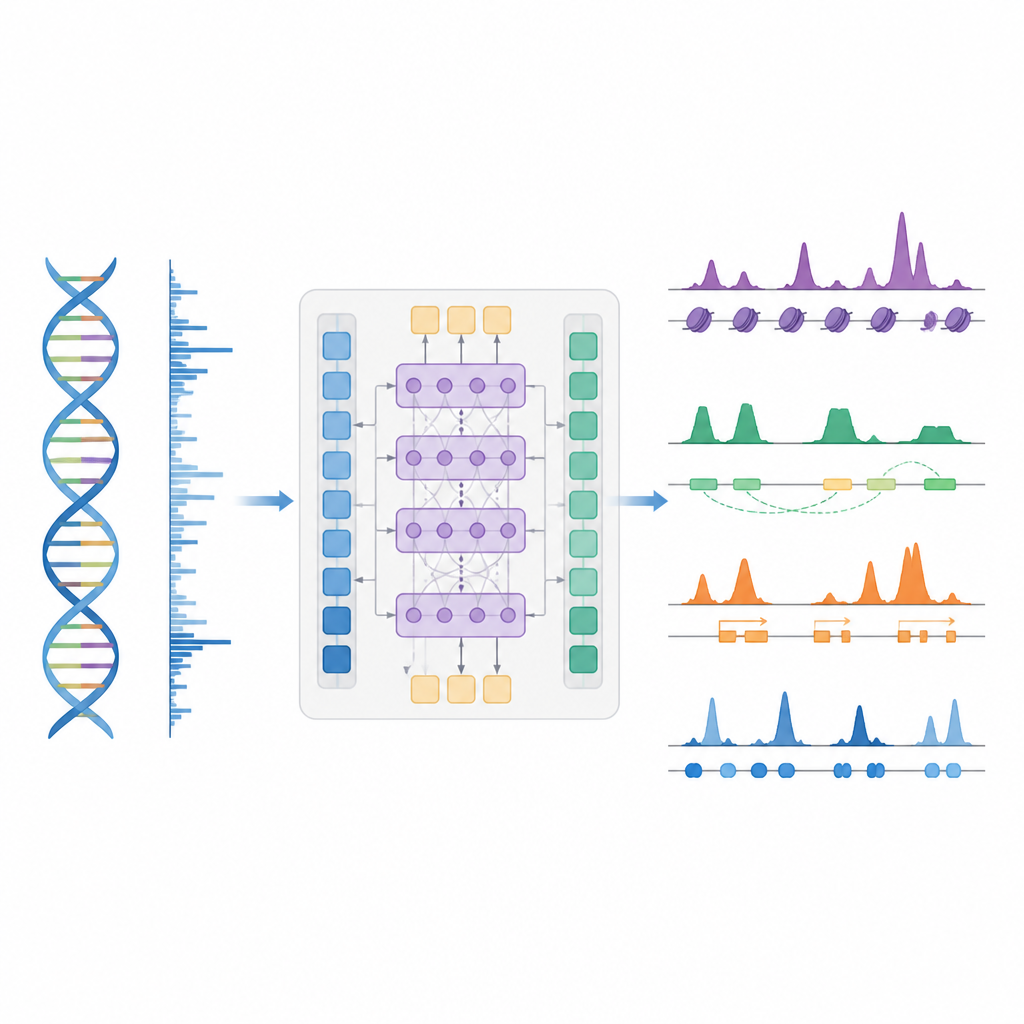
Sprytniejszy skrót do mapowania kontroli genów
Autorzy przedstawiają BioSeq2Seq, ramy głębokiego uczenia zaprojektowane do wnioskowania o różnych typach informacji regulatorowych na podstawie genomu. Zamiast powtarzać oddzielne eksperymenty dla każdej modyfikacji chemicznej czy białka, BioSeq2Seq uczy się na dwóch głównych wejściach. Pierwsze to sama sekwencja DNA, która jest taka sama w prawie wszystkich komórkach. Drugie to dane z asayu run-on, które raportują, gdzie polimeraza RNA aktywnie przesuwa się po DNA i w jakim kierunku. Ten assay daje „żywe” zdjęcie części genomu używanych w konkretnym typie komórek. Poprzez połączenie tych dwóch źródeł, model potrafi przewidzieć szeroki zakres cech, które biologowie zwykle mierzą oddzielnymi eksperymentami.
Jak model dostrzega wzorce w genomie
BioSeq2Seq opiera się na architekturze transformera, rodzaju sieci neuronowej pierwotnie używanym w modelach językowych. Tutaj „językiem” jest sekwencja zasad DNA oraz wzór sygnałów transkrypcyjnych wzdłuż chromosomu. Model najpierw przekształca zarówno DNA, jak i sygnały run-on w cechy numeryczne, a następnie używa warstw atencji, które potrafią łączyć odległe miejsca na ponad 100 000 zasad DNA. Ta dalekosiężna perspektywa jest istotna, ponieważ elementy kontrolne, takie jak wzmacniacze (enhancery), mogą działać daleko od genów, które regulują. Z tych udanych wzorców model generuje gęsto rozmieszczone przewidywania wzdłuż genomu, takie jak miejsca pojawiania się oznak aktywujących lub wyciszających na histonach, miejsca startu i zakończenia transkrypcji oraz typowe miejsca wiązania konkretnych białek.
Testowanie w wielu komórkach, tkankach i gatunkach
Naukowcy trenowali BioSeq2Seq głównie na danych z jednej ludzkiej linii komórkowej nowotworu krwi, a następnie testowali go w wielu innych kontekstach. Obejmowały one kilka ludzkich typów komórek, wątrobę myszy i konia oraz jajniki muszki owocowej. W przypadku dziesięciu typów modyfikacji histonów przewidywania modelu dobrze odpowiadały pomiarom eksperymentalnym, szczególnie dla oznak związanych z aktywnymi genami. Model radził sobie także dobrze w rejonach wokół miejsc startu genów oraz w promotorach i enhancerach, gdzie kontrola genów jest najbardziej nasilona. W porównaniu z wcześniejszymi narzędziami wykorzystującymi prostsze modele statystyczne lub mniej typów danych, BioSeq2Seq poprawił trafność przewidywań modyfikacji histonów średnio o ponad 14 procent i zrobił to znacznie szybciej, przewidując wszystkie oznaki jednocześnie zamiast pojedynczo.
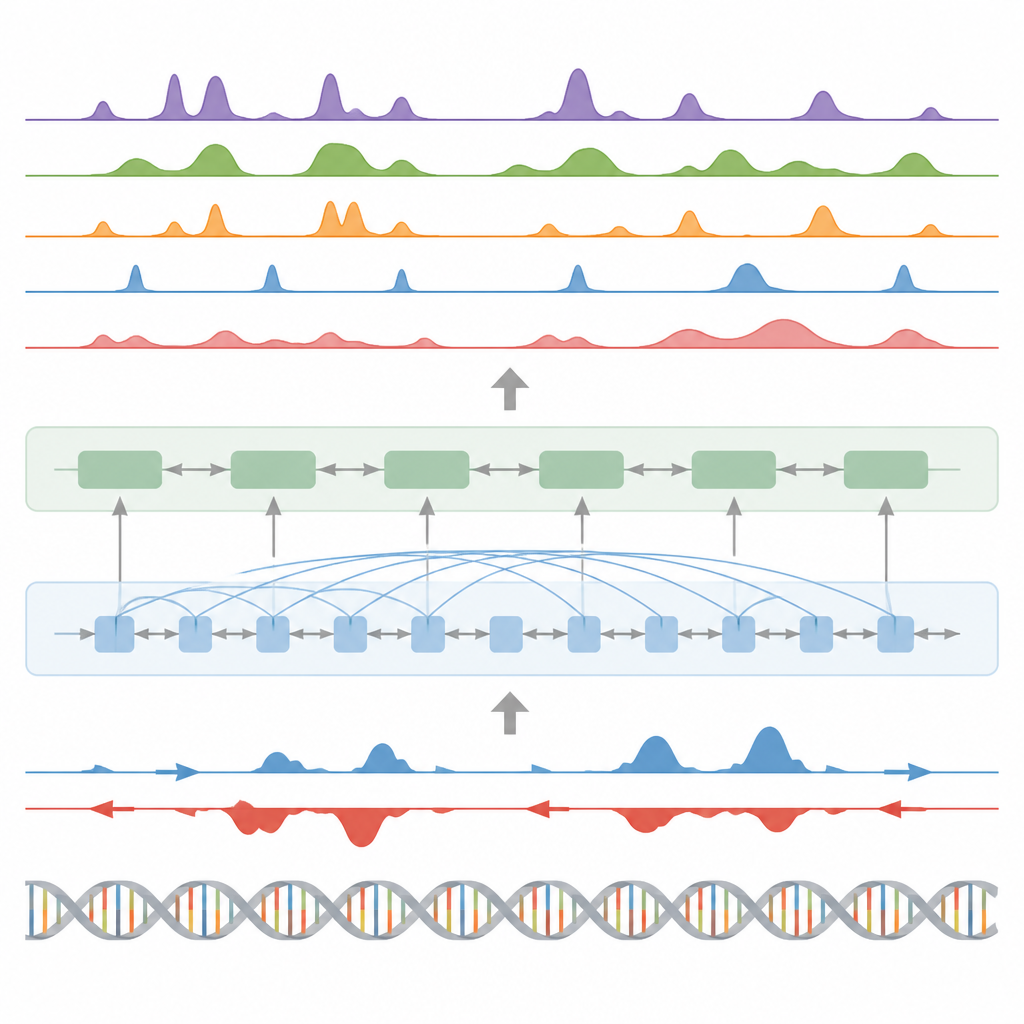
Wyszukiwanie kluczowych przełączników, aktywności genów i śladów białkowych
Ponad modyfikacjami histonów, model przetestowano w trzech innych głównych zadaniach. Po pierwsze, identyfikował elementy funkcjonalne, takie jak regiony startu transkrypcji, izolatory, miejsca poliadenylacji (poly(A)) oraz całe ciało genu, przekształcając ciągłe przewidywania sygnału w piki za pomocą niestandardowego detektora pików statystycznych. Dla regionów startu i ciał genów osiągnął wysokie wyniki zarówno pod względem dokładności, jak i odzysku (recall), przewyższając szeroko stosowaną metodę wykrywania aktywnych miejsc regulatorowych. Po drugie, BioSeq2Seq przewidywał pełne profile ekspresji genów, a nie tylko wysoki kontra niski poziom; prosty klasyfikator zbudowany na jego wyjściach pokonał kilka wiodących modeli, które polegają na znacznie większej liczbie eksperymentalnych danych. Po trzecie, używając tej samej ramy, autorzy nauczyli system przewidywania miejsc wiązania dla dziewięćdziesięciu różnych czynników transkrypcyjnych, osiągając wydajność zbliżoną do wiodącej metody wykorzystującej dane o otwartości chromatyny, a nawet poprawiając wyniki dla najtrudniejszych czynników przy użyciu jednego wspólnego modelu.
Co to oznacza dla badań genomów
Ucząc się, jak sekwencja DNA i pojedynczy assay transkrypcyjny odnoszą się do wielu warstw kontroli genów, BioSeq2Seq oferuje praktyczną alternatywę dla wykonywania dziesiątek oddzielnych eksperymentów. Pozwala badaczom wnioskować o modyfikacjach histonów, elementach regulatorowych, aktywności genów i wiązaniu białek w nowych typach komórek, tkankach, a nawet gatunkach, gdzie dostępne są jedynie dane run-on i referencyjny genom. Dla czytelnika niebędącego specjalistą kluczowa wiadomość jest taka, że jedno starannie wybrane doświadczenie, w połączeniu z potężnym systemem uczącym, może dziś zastąpić cały zestaw kosztownych testów, udostępniając duże badania regulacji genów dla znacznie większej liczby laboratoriów i pytań biologicznych.
Cytowanie: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
Słowa kluczowe: regulacja genów, głębokie uczenie, adnotacja genomu, transkrypcja, epigenomika