Clear Sky Science · en
An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation
Reading DNA without running every lab test
Modern biology often needs dozens of costly lab experiments to map how our genes are controlled in each cell type. This study shows how a single clever combination of sequencing data and artificial intelligence can stand in for many of those tests, offering a faster and cheaper way to read the control system of the genome.
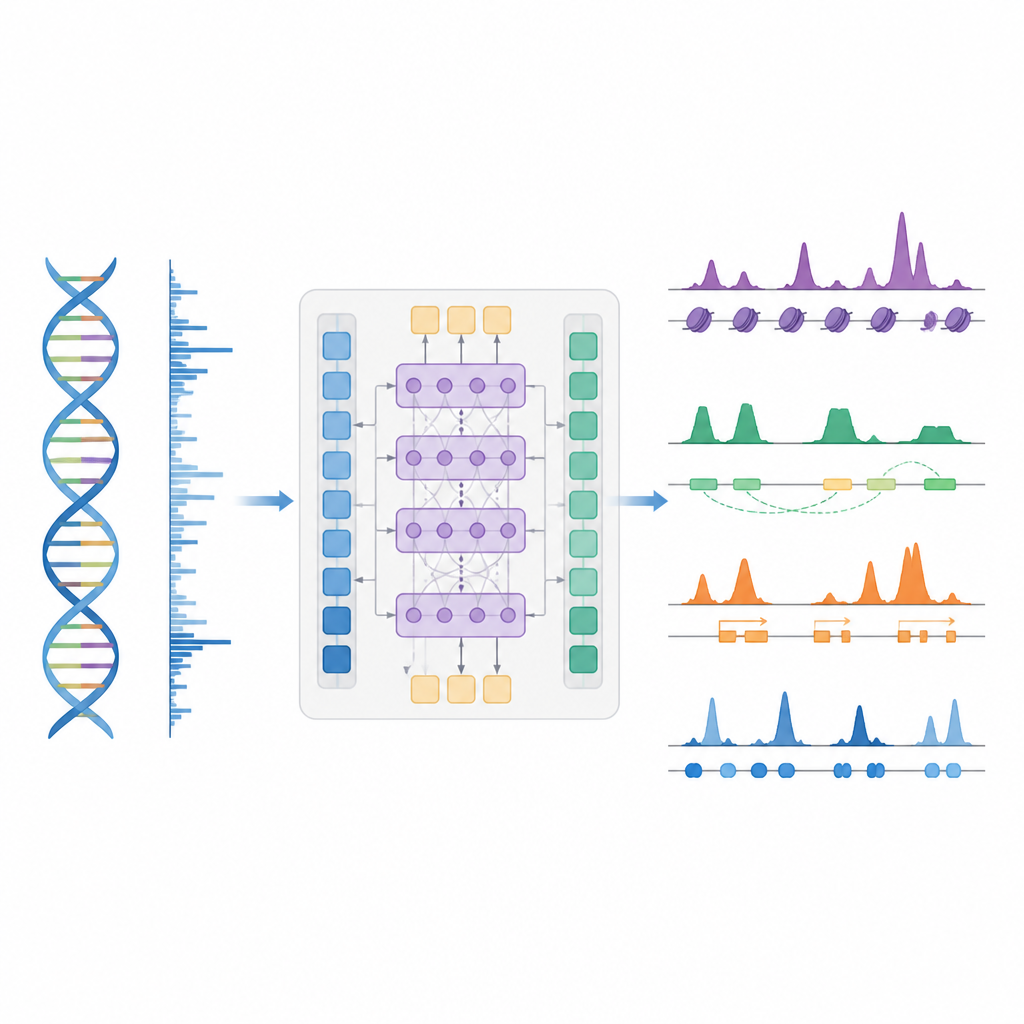
A smarter shortcut to map gene control
The authors introduce BioSeq2Seq, a deep learning framework designed to infer many kinds of regulatory information from the genome. Instead of repeating separate experiments for every chemical tag or protein, BioSeq2Seq learns from two main inputs. One is the DNA sequence itself, which is the same in almost all cells. The other is data from a run-on sequencing assay that reports where RNA polymerase is actively moving along DNA and in which direction. This assay captures a live snapshot of which parts of the genome are being used in a specific cell type. By combining these two sources, the model can predict a wide range of features that biologists usually measure with separate experiments.
How the model sees patterns in the genome
BioSeq2Seq relies on a transformer architecture, a type of neural network originally used in language models. Here, the “language” is the sequence of bases in DNA plus the pattern of transcription signals along the chromosome. The model first converts both DNA and run-on signals into numerical features and then uses attention layers that can connect distant sites across more than 100,000 DNA letters. This long view is important because control elements such as enhancers can act far from the genes they regulate. From these learned patterns, the model outputs finely spaced predictions along the genome, such as where activating or silencing tags on histone proteins should appear, where transcription starts or stops, and where specific proteins tend to bind.
Testing across many cells, tissues, and species
The researchers trained BioSeq2Seq mostly on data from one human blood cancer cell line, then challenged it on many other contexts. These included several human cell types, mouse and horse liver, and fruit fly ovary. Across ten types of histone marks, the model’s predictions matched experimental measurements closely, especially for marks linked to active genes. It also performed well in regions around gene start sites and within promoters and enhancers, where gene control is most intense. Compared with earlier tools that used simpler statistical models or fewer data types, BioSeq2Seq improved accuracy for histone marks by more than 14 percent on average and did so much faster, predicting all marks at once instead of one at a time.
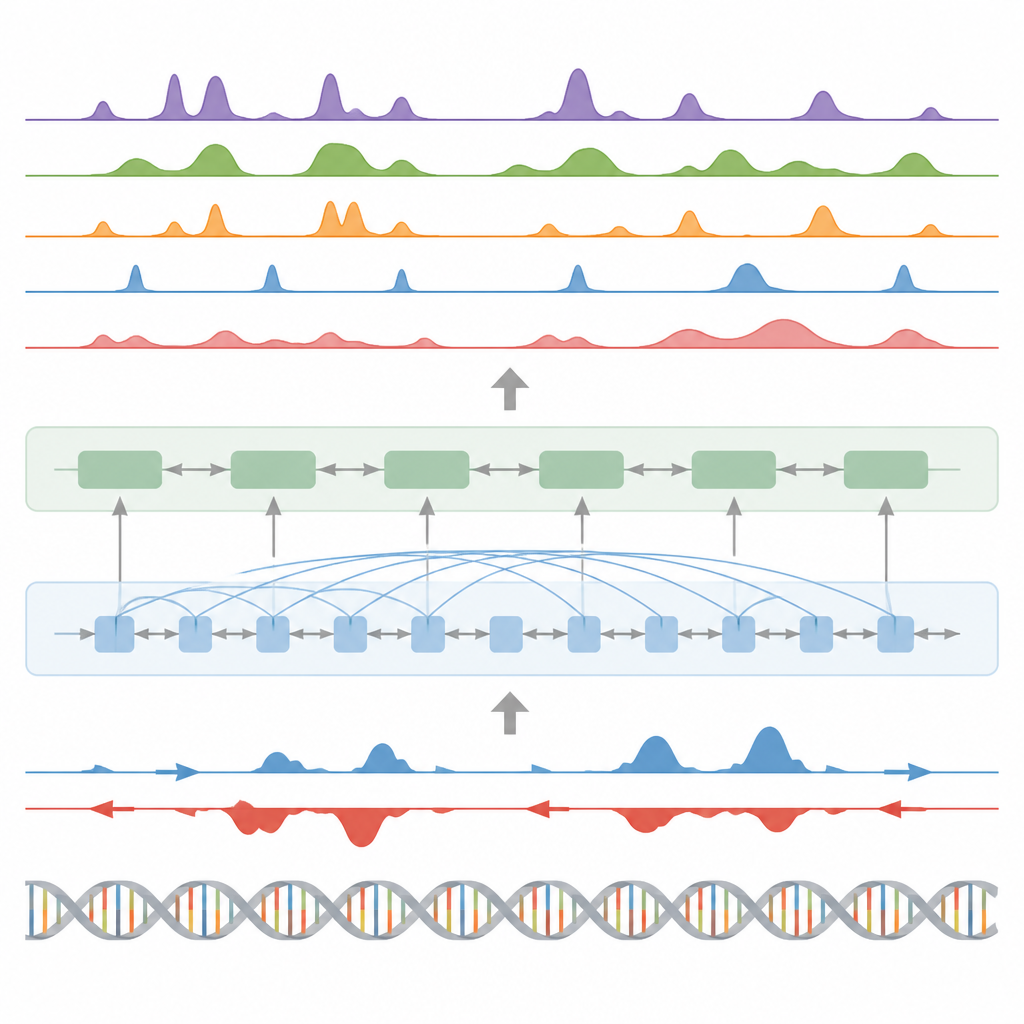
Finding key switches, gene activity, and protein footprints
Beyond histone marks, the model was tested on three other major tasks. First, it identified functional elements such as transcription start regions, insulators, poly(A) sites, and entire gene bodies by turning its continuous signal predictions into peaks with a custom statistical peak caller. For start regions and gene bodies, it reached high scores for both accuracy and recall and outperformed a widely used method for finding active regulatory sites. Second, BioSeq2Seq predicted full gene expression profiles, not just high versus low, and then a simple classifier built on its outputs beat several leading models that rely on many more experimental inputs. Third, using the same framework, the authors trained the system to predict binding sites for ninety different transcription factors, reaching performance similar to a top method that uses open-chromatin data and even improving on the hardest factors while using a single shared model.
What this means for studying genomes
By learning how DNA sequence and a single transcription assay relate to many layers of gene control, BioSeq2Seq offers a practical alternative to running dozens of separate experiments. It allows researchers to infer histone marks, regulatory elements, gene activity, and protein binding in new cell types, tissues, and even species where only run-on data and a reference genome are available. For a lay reader, the key message is that one carefully chosen experiment, combined with a powerful learning system, can now stand in for an entire toolbox of costly tests, bringing large-scale studies of gene regulation within reach for many more labs and biological questions.
Citation: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
Keywords: gene regulation, deep learning, genome annotation, transcription, epigenomics