Clear Sky Science · pt
Uma estrutura de deep learning generalizável de ponta a ponta para analisar de forma abrangente a regulação transcricional
Lendo o DNA sem realizar todos os testes de laboratório
A biologia moderna frequentemente precisa de dezenas de experimentos laboratoriais caros para mapear como nossos genes são controlados em cada tipo celular. Este estudo demonstra como uma única combinação astuta de dados de sequenciamento e inteligência artificial pode substituir muitos desses testes, oferecendo uma forma mais rápida e barata de interpretar o sistema de controle do genoma.
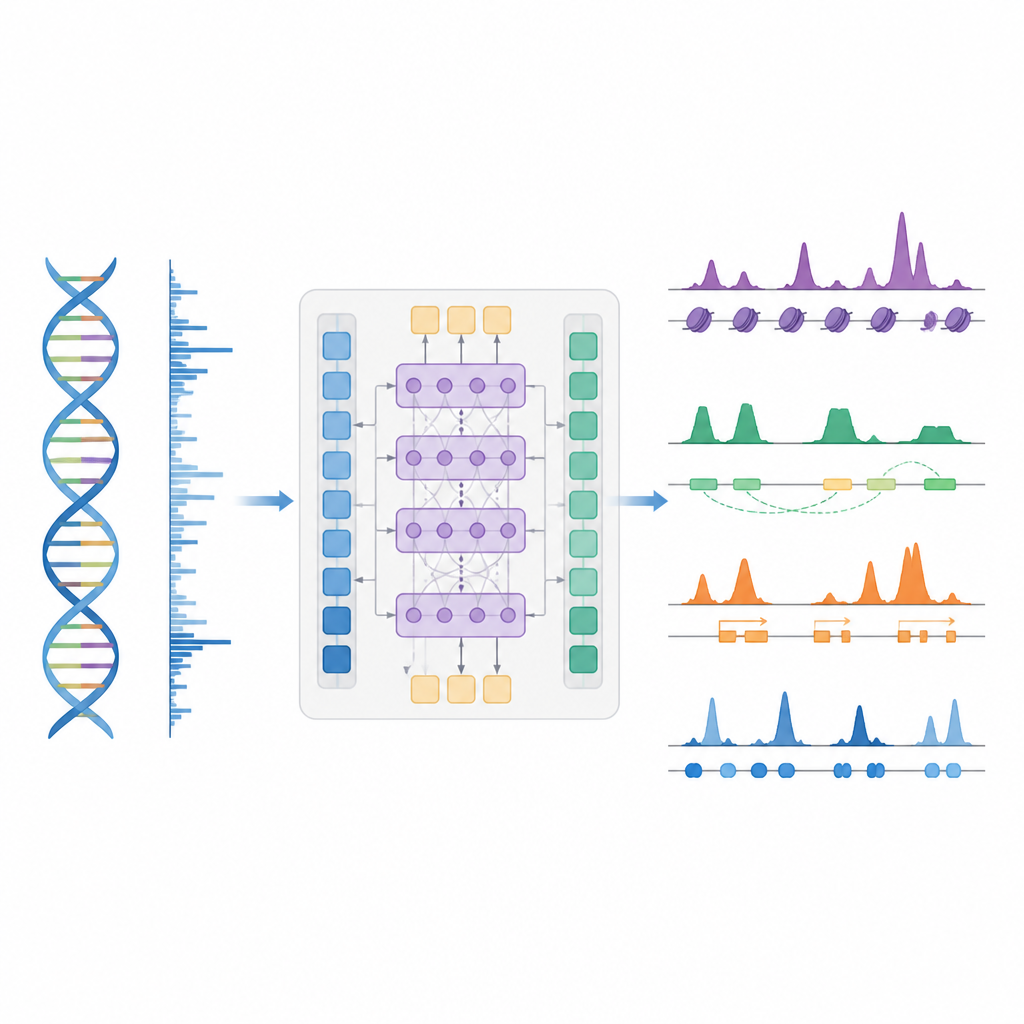
Um atalho mais inteligente para mapear o controle gênico
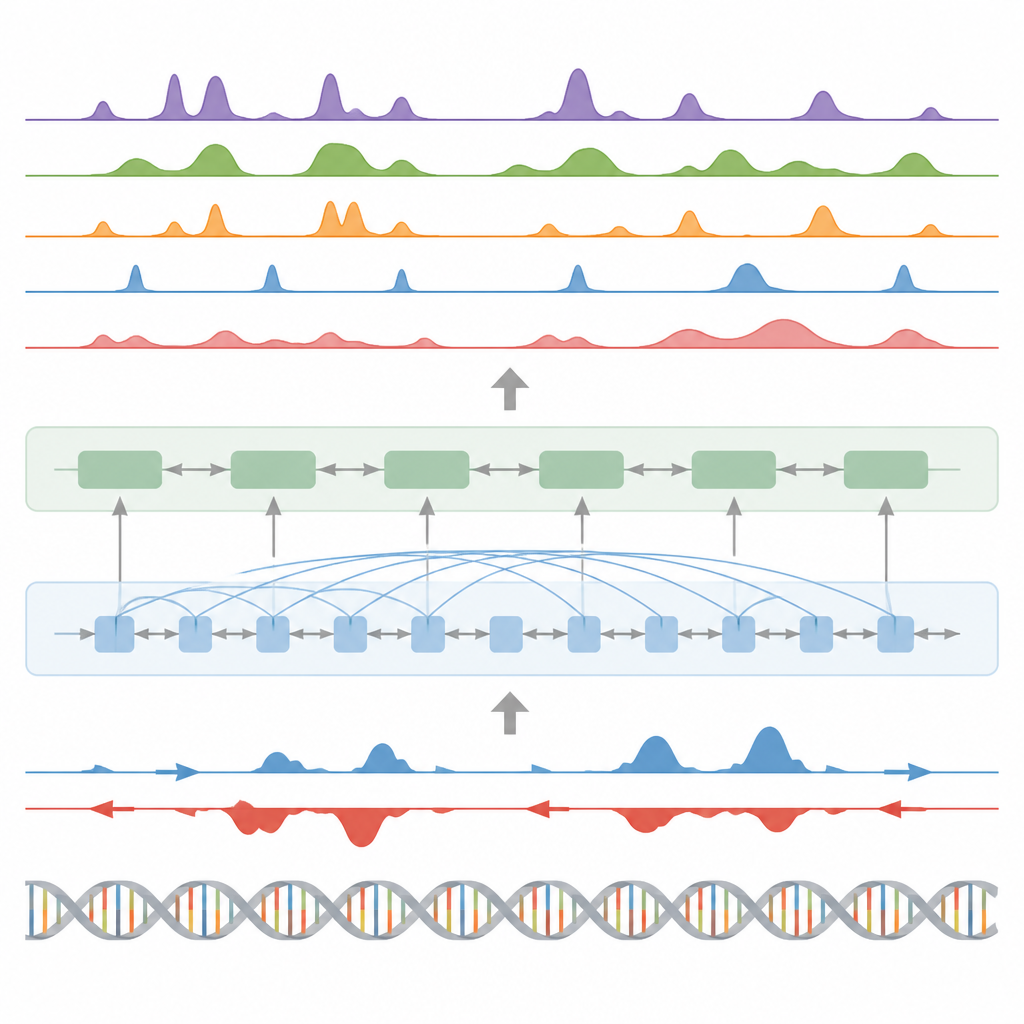
Os autores apresentam o BioSeq2Seq, uma estrutura de deep learning projetada para inferir muitos tipos de informação regulatória a partir do genoma. Em vez de repetir experimentos separados para cada marca química ou proteína, o BioSeq2Seq aprende a partir de duas entradas principais. Uma é a própria sequência de DNA, que é a mesma na maior parte das células. A outra são dados de um ensaio de sequenciamento run-on que informa onde a RNA polimerase está se movendo ativamente ao longo do DNA e em que direção. Esse ensaio captura um instantâneo em tempo real de quais partes do genoma estão sendo utilizadas em um tipo celular específico. Ao combinar essas duas fontes, o modelo pode prever uma ampla gama de características que biólogos normalmente medem com experimentos separados.
Como o modelo enxerga padrões no genoma
O BioSeq2Seq baseia-se em uma arquitetura transformer, um tipo de rede neural originalmente usada em modelos de linguagem. Aqui, a “linguagem” é a sequência de bases do DNA mais o padrão de sinais de transcrição ao longo do cromossomo. O modelo primeiro converte tanto o DNA quanto os sinais run-on em características numéricas e então usa camadas de atenção que podem conectar sítios distantes ao longo de mais de 100.000 letras de DNA. Essa visão de longo alcance é importante porque elementos de controle, como enhancers, podem atuar longe dos genes que regulam. A partir desses padrões aprendidos, o modelo produz previsões finamente distribuídas ao longo do genoma, como onde devem aparecer marcas ativadoras ou de silenciamento em histonas, onde a transcrição começa ou termina e onde proteínas específicas tendem a se ligar.
Testes em muitas células, tecidos e espécies
Os pesquisadores treinaram o BioSeq2Seq principalmente com dados de uma linha celular humana de câncer sanguíneo e depois o desafiaram em muitos outros contextos. Esses incluíram vários tipos celulares humanos, fígado de camundongo e cavalo e ovário da mosca-da-fruta. Em dez tipos de marcas de histonas, as previsões do modelo corresponderam de perto às medições experimentais, especialmente para marcas associadas a genes ativos. Ele também teve bom desempenho em regiões ao redor dos sítios de início de genes e dentro de promotores e enhancers, onde o controle gênico é mais intenso. Em comparação com ferramentas anteriores que usavam modelos estatísticos mais simples ou menos tipos de dados, o BioSeq2Seq melhorou a acurácia para marcas de histonas em mais de 14% em média e fez isso muito mais rápido, prevendo todas as marcas de uma vez em vez de uma a uma.
Encontrando interruptores chave, atividade gênica e pegadas de proteínas
Além das marcas de histonas, o modelo foi testado em três outras tarefas principais. Primeiro, ele identificou elementos funcionais como regiões de início de transcrição, isoladores, sítios de poliadenilação (poly(A)) e corpos gênicos inteiros transformando suas previsões contínuas em picos com um detector de picos estatístico customizado. Para regiões de início e corpos gênicos, alcançou pontuações altas tanto em precisão quanto em recall e superou um método amplamente utilizado para encontrar sítios regulatórios ativos. Segundo, o BioSeq2Seq previu perfis completos de expressão gênica, não apenas alto versus baixo, e então um classificador simples construído sobre suas saídas superou vários modelos líderes que dependem de muitos mais insumos experimentais. Terceiro, usando a mesma estrutura, os autores treinaram o sistema para prever sítios de ligação de noventa diferentes fatores de transcrição, atingindo desempenho similar a um método de ponta que usa dados de cromatina aberta e até melhorando nos fatores mais difíceis, enquanto utiliza um único modelo compartilhado.
O que isso significa para o estudo dos genomas
Ao aprender como a sequência de DNA e um único ensaio de transcrição se relacionam com muitas camadas de controle gênico, o BioSeq2Seq oferece uma alternativa prática a realizar dezenas de experimentos separados. Ele permite que pesquisadores inferam marcas de histonas, elementos regulatórios, atividade gênica e ligação de proteínas em novos tipos celulares, tecidos e até espécies onde apenas dados run-on e um genoma de referência estão disponíveis. Para um leitor leigo, a mensagem principal é que um experimento cuidadosamente escolhido, combinado com um sistema de aprendizado poderoso, pode agora substituir um conjunto inteiro de testes caros, aproximando estudos em larga escala da regulação gênica para muito mais laboratórios e questões biológicas.
Citação: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
Palavras-chave: regulação gênica, deep learning, anotação do genoma, transcrição, epigenômica