Clear Sky Science · fr
Un cadre d’apprentissage profond généralisable de bout en bout pour analyser de manière exhaustive la régulation transcriptionnelle
Lire l’ADN sans réaliser chaque test de laboratoire
La biologie moderne nécessite souvent des dizaines d’expériences de laboratoire coûteuses pour cartographier la manière dont nos gènes sont contrôlés dans chaque type cellulaire. Cette étude montre comment une unique combinaison astucieuse de données de séquençage et d’intelligence artificielle peut remplacer nombre de ces tests, offrant une façon plus rapide et moins onéreuse de lire le système de contrôle du génome.
Un raccourci plus intelligent pour cartographier le contrôle des gènes
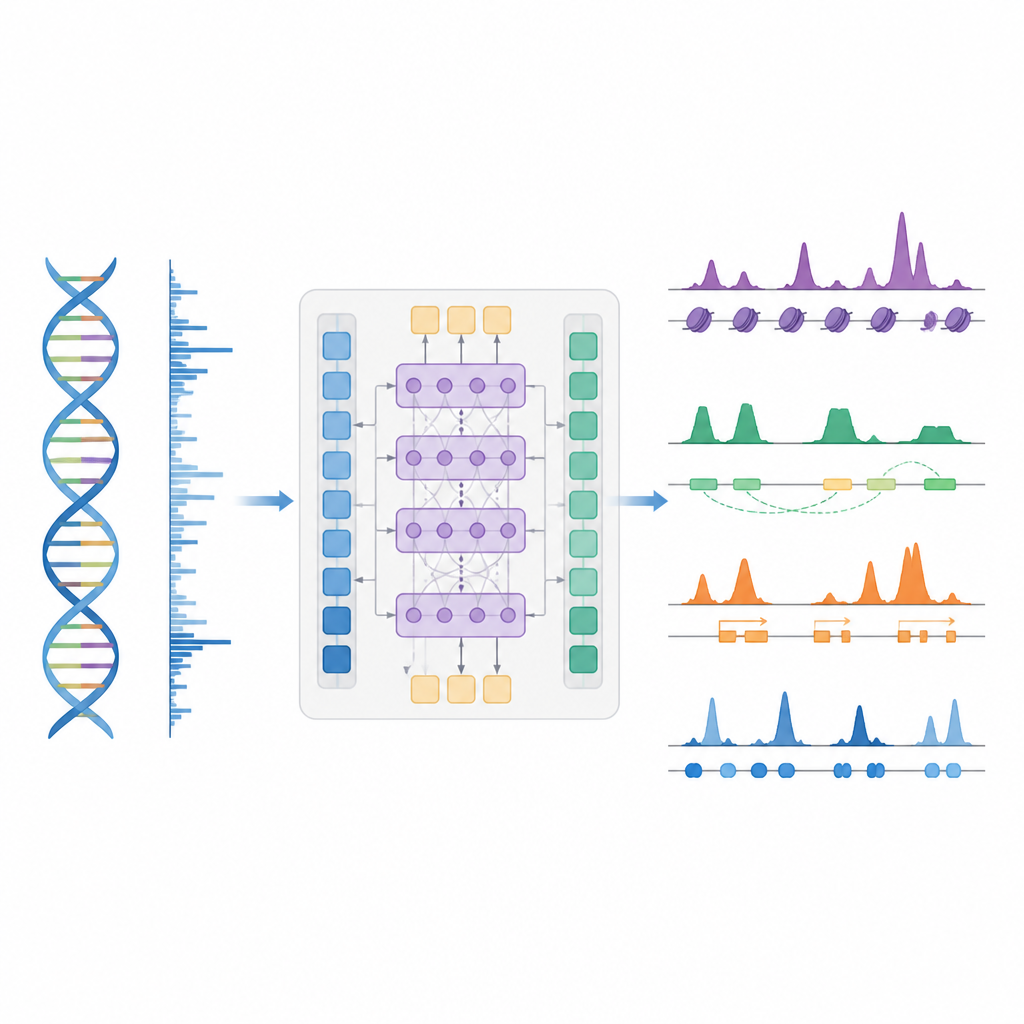
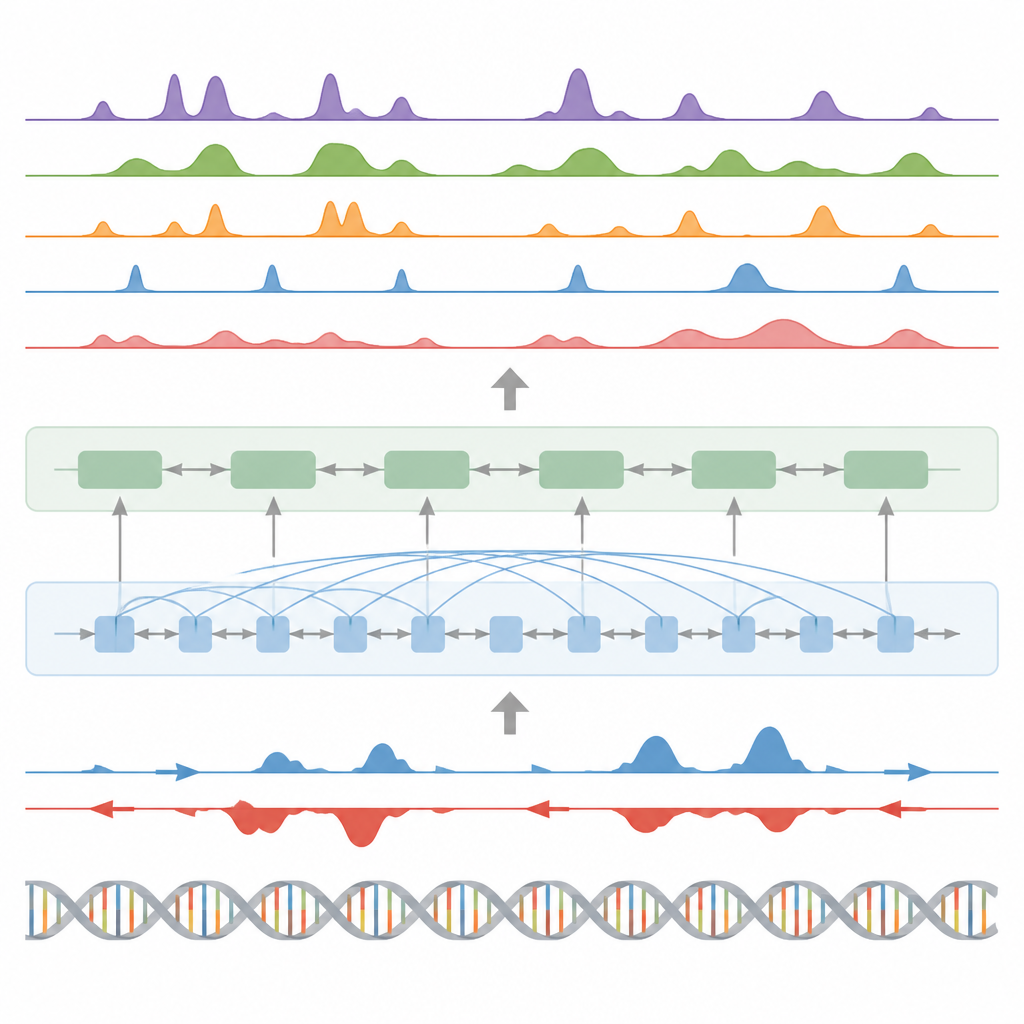
Les auteurs présentent BioSeq2Seq, un cadre d’apprentissage profond conçu pour inférer de nombreux types d’informations régulatrices à partir du génome. Plutôt que de répéter des expériences séparées pour chaque marque chimique ou protéine, BioSeq2Seq apprend à partir de deux entrées principales. L’une est la séquence d’ADN elle‑même, qui est la même dans presque toutes les cellules. L’autre est constituée des données d’un test de séquençage run‑on qui indique où l’ARN polymérase se déplace activement le long de l’ADN et dans quelle direction. Cet essai capture un instantané en direct des régions du génome utilisées dans un type cellulaire donné. En combinant ces deux sources, le modèle peut prédire un large éventail de caractéristiques que les biologistes mesurent habituellement avec des expériences distinctes.
Comment le modèle détecte les motifs dans le génome
BioSeq2Seq s’appuie sur une architecture de type transformer, un type de réseau neuronal initialement utilisé dans les modèles de langage. Ici, la « langue » est la séquence de bases de l’ADN plus le motif des signaux de transcription le long du chromosome. Le modèle convertit d’abord l’ADN et les signaux run‑on en caractéristiques numériques, puis utilise des couches d’attention capables de relier des sites distants sur plus de 100 000 nucléotides. Cette vue à longue portée est importante parce que des éléments de contrôle tels que les enhancers peuvent agir loin des gènes qu’ils régulent. À partir de ces motifs appris, le modèle fournit des prédictions finement spatialisées le long du génome, comme l’emplacement des marques d’activation ou de répression sur les histones, les sites de démarrage ou d’arrêt de la transcription, et les endroits où des protéines spécifiques ont tendance à se lier.
Test sur de nombreuses cellules, tissus et espèces
Les chercheurs ont entraîné BioSeq2Seq principalement sur des données issues d’une lignée cellulaire humaine de cancer du sang, puis l’ont testé dans de nombreux autres contextes. Ceux‑ci incluaient plusieurs types cellulaires humains, le foie de souris et de cheval, et l’ovaire de drosophile. Pour dix types de marques d’histones, les prédictions du modèle concordaient étroitement avec les mesures expérimentales, en particulier pour les marques associées aux gènes actifs. Il a également donné de bonnes performances dans les régions autour des sites de démarrage des gènes et à l’intérieur des promoteurs et enhancers, où le contrôle génique est le plus intense. Comparé aux outils antérieurs qui utilisaient des modèles statistiques plus simples ou moins de types de données, BioSeq2Seq a amélioré la précision pour les marques d’histones de plus de 14 % en moyenne et l’a fait bien plus rapidement, en prédisant toutes les marques simultanément plutôt qu’une par une.
Identifier les interrupteurs clés, l’activité génique et les empreintes protéiques
Au‑delà des marques d’histones, le modèle a été évalué sur trois autres tâches majeures. Premièrement, il a identifié des éléments fonctionnels tels que les régions de démarrage de la transcription, les isolateurs, les sites poly(A) et les corps de gènes entiers en transformant ses prédictions de signal continu en pics à l’aide d’un détecteur de pics statistique personnalisé. Pour les régions de démarrage et les corps de gènes, il a atteint des scores élevés en précision et en rappel et a surpassé une méthode largement utilisée pour la détection de sites régulateurs actifs. Deuxièmement, BioSeq2Seq a prédit des profils d’expression génique complets, pas seulement haut vs bas, et un classifieur simple construit sur ses sorties a battu plusieurs modèles de pointe qui s’appuient sur beaucoup plus d’entrées expérimentales. Troisièmement, en utilisant le même cadre, les auteurs ont entraîné le système à prédire les sites de liaison pour quatre‑vingt‑dix facteurs de transcription différents, atteignant une performance comparable à une méthode de référence qui utilise des données d’accessibilité de la chromatine et améliorant même les prédictions pour les facteurs les plus difficiles tout en utilisant un seul modèle partagé.
Ce que cela implique pour l’étude des génomes
En apprenant comment la séquence d’ADN et un seul essai de transcription se rapportent à de nombreuses couches de contrôle génique, BioSeq2Seq offre une alternative pratique à la réalisation de dizaines d’expériences distinctes. Il permet aux chercheurs d’inférer des marques d’histones, des éléments régulateurs, l’activité génique et la liaison des protéines dans de nouveaux types cellulaires, tissus et même espèces où seules des données run‑on et un génome de référence sont disponibles. Pour le lecteur non spécialiste, le message clé est qu’une expérience soigneusement choisie, combinée à un système d’apprentissage puissant, peut désormais remplacer un ensemble entier de tests coûteux, rapprochant ainsi les études à grande échelle de la régulation génique pour beaucoup plus de laboratoires et de questions biologiques.
Citation: Zhang, Z., Fan, X., Zhong, J. et al. An end-to-end generalizable deep learning framework to comprehensively analyze transcriptional regulation. Nat Commun 17, 4708 (2026). https://doi.org/10.1038/s41467-026-70070-6
Mots-clés: régulation des gènes, apprentissage profond, annotation du génome, transcription, épigénomique