Clear Sky Science · zh
BCR::ABL1 酪氨酸激酶抑制剂通过引发核糖体碰撞激活依赖 ZAK 的核糖毒性应激并在慢性髓性白血病中诱导凋亡
当抗癌药与细胞的蛋白质工厂对话时
慢性髓性白血病(CML)通常通过靶向药物控制,这些药物能关闭一种名为 BCR::ABL1 的促癌酶。但部分患者仍会进展到侵袭性阶段或对治疗产生耐药性。本研究揭示了一个出人意料的因素,参与这些药物杀伤癌细胞的过程:细胞自身的蛋白质合成机器——核糖体。通过观察这些机器堵塞和碰撞时的变化,作者发现了一条应激通路,这条通路既有助于 CML 细胞生长,又在药物治疗下促使它们被消灭。
血液癌细胞内的隐秘应激警报
核糖体通常像节奏良好的装配线,读取遗传信息并缝合出蛋白质。然而在应激下,核糖体会发生停滞并相互碰撞,从而触发细胞内的“核糖毒性”报警系统。研究人员将注意力集中在一个位于应激信号链顶端的感受蛋白 ZAK 上。通过分析近 200 例 CML 患者的样本,他们发现 ZAK 和若干相关的质量控制因子在进展到危险的爆发期(blast phase)的患者中明显增多,且更高的 ZAK 水平与癌性未成熟血细胞比例增高相关。这表明随着 CML 恶化,其细胞越来越依赖与 ZAK 相关的通路。
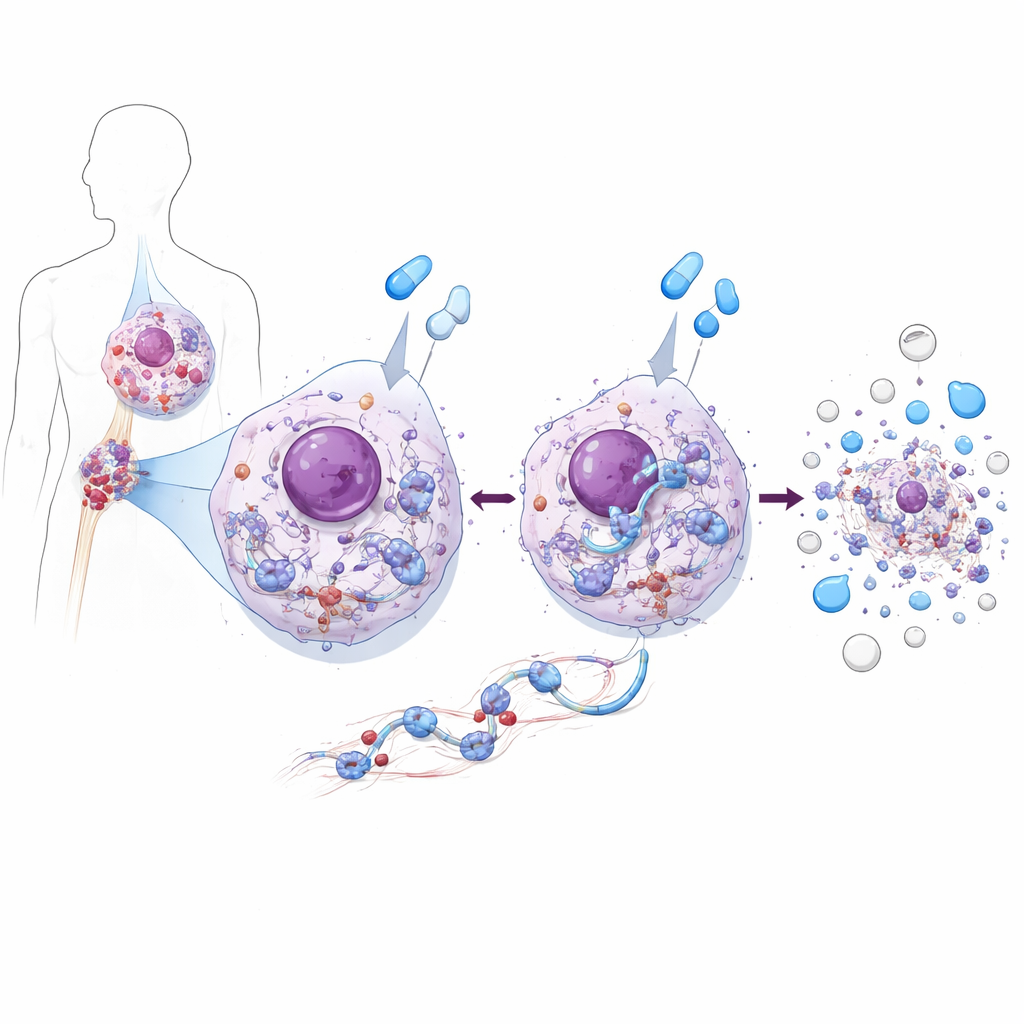
既养护又致命的一个蛋白
在 CML 细胞系和患者来源细胞中,研究团队发现 ZAK 具有双重性格。在未治疗的 CML 细胞中,ZAK 支持细胞生长。它增强了一个主要促生长通路——AKT–mTOR 通路的活性,进而驱动蛋白质生成和细胞分裂。当去除或抑制 ZAK 时,白血病细胞的分裂减慢,因为该促生长通路被削弱。但当同样的细胞接受如伊马替尼(imatinib)或 asciminib 之类的 BCR::ABL1 阻断药物处理时,ZAK 则扮演相反角色:它是这些药物触发高效细胞死亡所必需的。没有 ZAK,这些药物虽然仍能抑制其靶酶,但白血病细胞发生凋亡的可能性大大降低,即便是在直接取自 CML 患者的样本中也是如此。
药物减速的蛋白质合成如何引发核糖体堆积
作者接着探究关闭 BCR::ABL1 如何导致 ZAK 被激活。他们发现 BCR::ABL1 正常情况下维持 mTOR 通路的开启,从而保持快速的蛋白质合成。药物阻断 BCR::ABL1 后,mTOR 活性下降。这一变化翻转了下游开关,减慢了核糖体沿 RNA 模板移动的速度。对新合成蛋白的测量证实了翻译速度减慢,专门的梯度实验显示药物处理的细胞中核糖体变得拥挤并形成易发生碰撞的簇。这些堵塞的核糖体对核酸酶的降解异常耐受,并富集有如 EDF1 和 ZAK 等碰撞传感器,清楚地表明细胞的蛋白质工厂承受了机械应力。
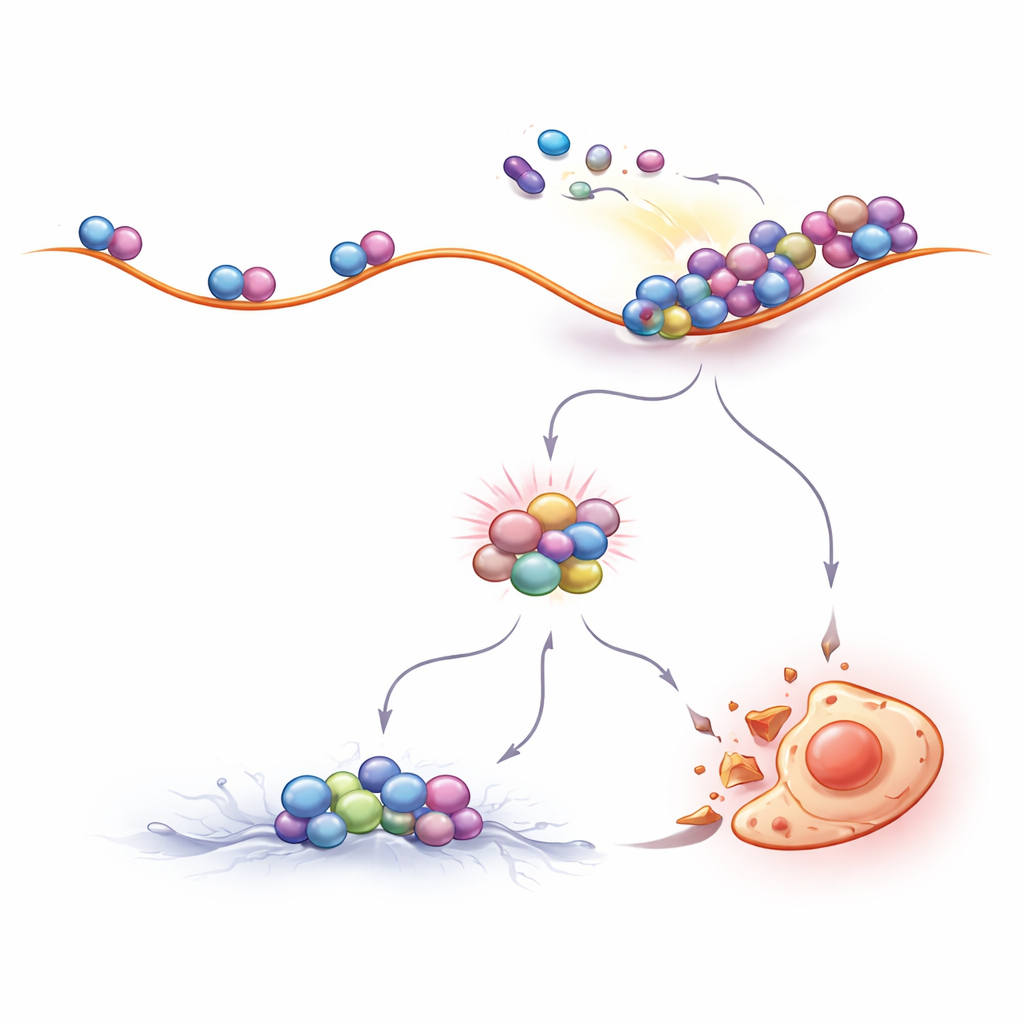
调节核糖体流量以提高治疗效果
如果核糖体碰撞是 CML 药物发挥作用的核心,那么有意改变核糖体的流量就应改变药物敏感性。研究者确实观察到了这一点。减少启动新蛋白链的核糖体数量的化合物降低了碰撞发生的可能性,从而保护 CML 细胞免受伊马替尼诱导的死亡。相反,略微减慢核糖体沿 RNA 逐步移动的药物,或在细胞内激活相同的减速机制,都会增强碰撞并显著提高 BCR::ABL1 抑制剂对 CML 细胞的杀伤效果。重要的是,这一效应不仅在实验室细胞系中观察到,也存在于来自患者的原代细胞中,强调了其潜在的临床相关性。
这对患者与未来治疗的意义
对非专业读者而言,关键观点是:CML 药物的作用不仅仅是关闭一个失控的酶;它们还会使癌细胞的蛋白质装配线发生堵塞,导致核糖体碰撞。这些碰撞唤醒 ZAK,使其从促进生长的角色转变为执行者,通过触发一条以细胞自杀为终点的应激通路来杀死细胞。通过学习如何精细调控核糖体的速度——在合适的方式下加速或减慢——医生未来或可增强现有 CML 药物的疗效或克服耐药。该研究表明,核糖体的物理行为,而不仅是它们产生蛋白质的数量,是白血病乃至其他癌症中一个强大且可药物化的弱点。
引用: Park, J., Kim, SH., Park, J. et al. BCR::ABL1 tyrosine kinase inhibitors induce ribosome collisions to activate ZAK-dependent ribotoxic stress and apoptosis in chronic myeloid leukemia. Leukemia 40, 955–969 (2026). https://doi.org/10.1038/s41375-026-02916-3
关键词: 慢性髓性白血病, 核糖体碰撞, ZAK 应激激酶, BCR-ABL 抑制剂, 蛋白质合成