Clear Sky Science · en
BCR::ABL1 tyrosine kinase inhibitors induce ribosome collisions to activate ZAK-dependent ribotoxic stress and apoptosis in chronic myeloid leukemia
When Cancer Drugs Talk to the Cell’s Protein Factories
Chronic myeloid leukemia (CML) is often held in check by targeted drugs that shut down a cancer‑driving enzyme called BCR::ABL1. Yet some patients still progress to an aggressive stage or become resistant to treatment. This study explores a surprising player in how these drugs kill cancer cells: the cell’s own protein‑making machines, the ribosomes. By watching what happens when these machines jam and collide, the authors uncover a stress pathway that both helps CML cells grow and, under drug treatment, helps destroy them.
A Hidden Stress Alarm Inside Blood Cancer Cells
Ribosomes usually work like well‑timed assembly lines, reading genetic messages and stitching together proteins. Under stress, however, ribosomes can stall and crash into one another, setting off a “ribotoxic” alarm system inside the cell. The researchers focused on a sensor protein called ZAK that sits at the top of a stress‑signaling chain. By analyzing samples from nearly 200 CML patients, they found that ZAK and several related quality‑control factors were much more abundant in patients whose disease had advanced to the dangerous blast phase, and that higher ZAK levels tracked with a higher proportion of immature cancerous blood cells. This suggested that as CML worsens, its cells lean heavily on ZAK‑linked pathways.
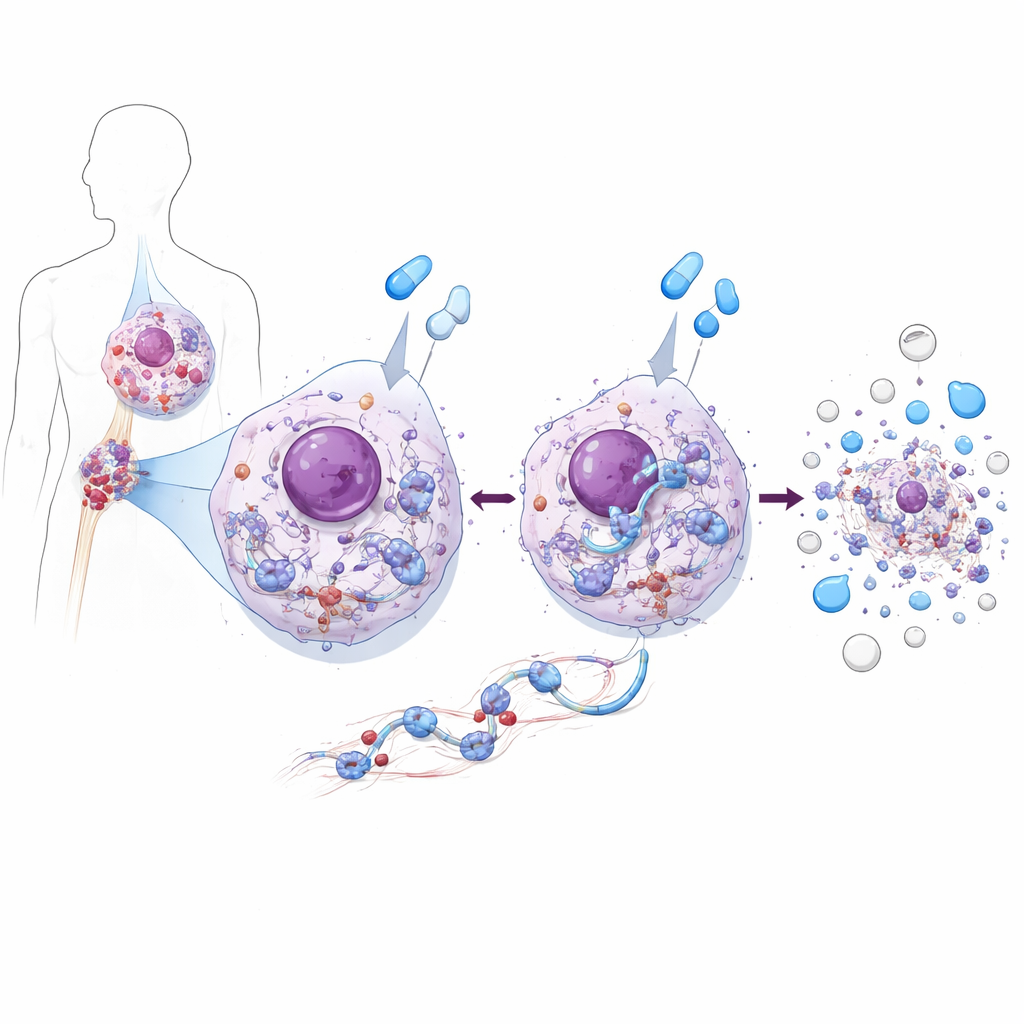
One Protein that Both Feeds and Kills Leukemia Cells
Using CML cell lines and patient‑derived cells, the team discovered that ZAK has a double personality. In untreated CML cells, ZAK supports growth. It boosts activity in a major growth‑promoting chain, the AKT–mTOR pathway, which in turn drives protein production and cell division. When ZAK was removed or blocked, leukemia cells divided more slowly because this growth pathway weakened. But when the same cells were treated with BCR::ABL1‑blocking drugs such as imatinib or asciminib, ZAK played the opposite role: it was required for the drugs to trigger efficient cell death. Without ZAK, these drugs still hit their target enzyme but the leukemia cells were much less likely to undergo apoptosis, even in samples taken directly from CML patients.
How Drug‑Slowed Protein Making Causes Ribosome Pileups
The authors then asked how shutting off BCR::ABL1 leads to ZAK activation. They found that BCR::ABL1 normally keeps the mTOR pathway switched on, which maintains fast protein production. When the drugs blocked BCR::ABL1, mTOR activity dropped. This change flipped downstream switches that slow the movement of ribosomes along their RNA templates. Measurements of newly made proteins confirmed that translation slowed, and specialized gradient experiments showed that ribosomes in drug‑treated cells became crowded and formed collision‑prone clusters. These jammed ribosomes were unusually resistant to breakdown by nucleases and were loaded with collision sensors such as EDF1 and ZAK, clear signatures that the cell’s protein factories were under mechanical stress.
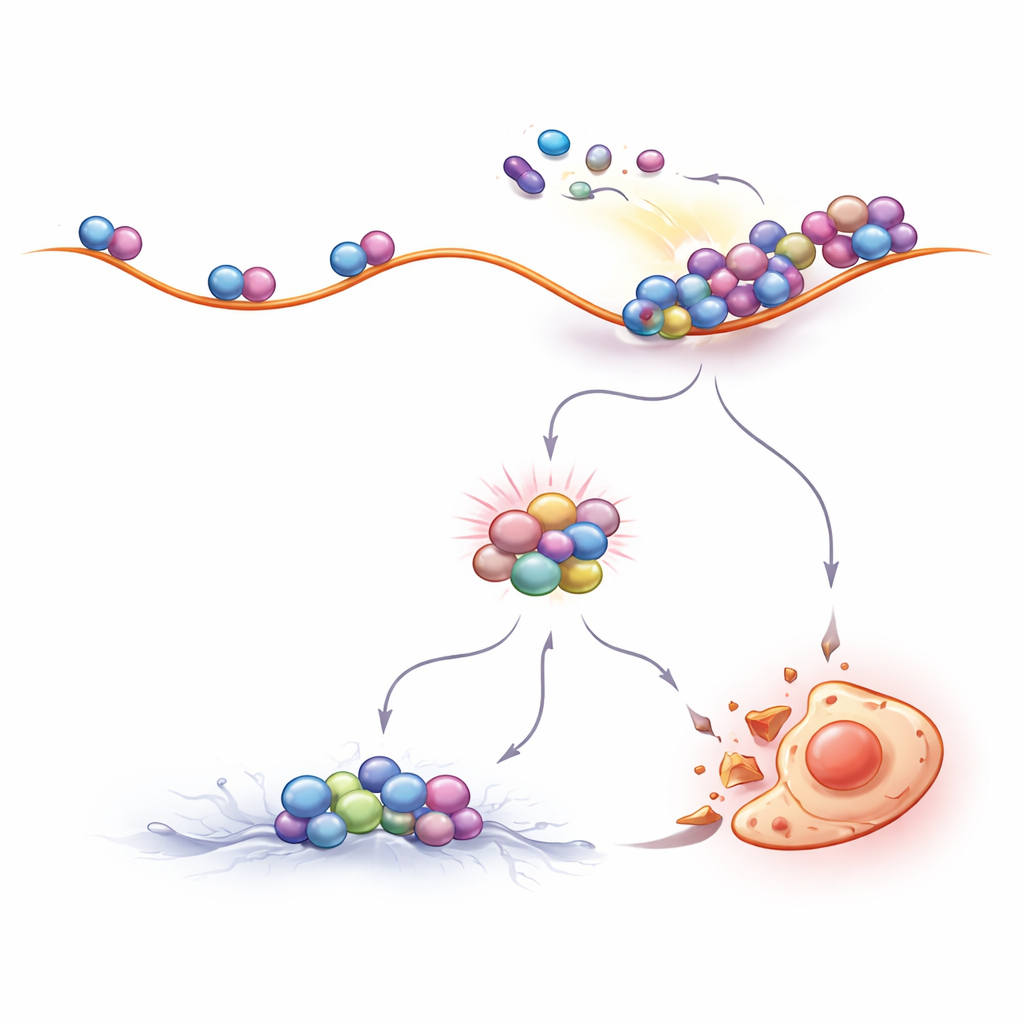
Tuning Ribosome Traffic to Make Treatment Work Better
If ribosome collisions are central to how CML drugs work, then deliberately changing ribosome traffic should alter drug sensitivity. This is exactly what the researchers observed. Compounds that reduced the number of ribosomes starting new protein chains made collisions less likely and protected CML cells from imatinib‑induced death. In contrast, drugs that slightly slowed the step‑by‑step movement of ribosomes along RNA, or that activated the same slowing mechanism inside the cell, boosted collisions and greatly enhanced killing of CML cells by BCR::ABL1 inhibitors. Importantly, this effect was seen not only in laboratory cell lines but also in primary cells taken from patients, underscoring its potential clinical relevance.
What This Means for Patients and Future Therapies
To a non‑specialist, the key message is that CML drugs do more than just turn off a rogue enzyme; they also jam the cancer cell’s protein assembly lines, causing ribosomes to collide. These collisions awaken ZAK, which flips from being a growth helper to acting as an executioner by triggering a stress pathway that ends in cell suicide. By learning how to fine‑tune ribosome speed—speeding it up or slowing it down in the right way—doctors may eventually boost the effectiveness of existing CML drugs or overcome resistance. The study suggests that the physical behavior of ribosomes, not just the amount of protein they make, is a powerful and druggable vulnerability in leukemia and potentially other cancers.
Citation: Park, J., Kim, SH., Park, J. et al. BCR::ABL1 tyrosine kinase inhibitors induce ribosome collisions to activate ZAK-dependent ribotoxic stress and apoptosis in chronic myeloid leukemia. Leukemia 40, 955–969 (2026). https://doi.org/10.1038/s41375-026-02916-3
Keywords: chronic myeloid leukemia, ribosome collisions, ZAK stress kinase, BCR-ABL inhibitors, protein synthesis