Clear Sky Science · sv
Riktlinjer för att optimera referenspaneler för DNA-metylering vid cellulär dekonvolution
Att skåda in i blandade vävnader
Nutida studier av hälsa och sjukdom mäter ofta kemiska markörer på vårt DNA för att förstå hur miljö och livsstil lämnar spår i våra gener. Men de flesta analyser görs på blandade vävnader, som blod, som innehåller många olika celltyper. Om vi inte kan avgöra hur stor andel av varje celltyp som finns, kan vi missta en förändring i cellblandningen för en verklig sjukdomssignal. Denna artikel förklarar hur man bygger bättre ”referenspaneler” som gör det möjligt för forskare att exakt uppskatta cellblandningar från DNA-metyleringsdata, vilket leder till tydligare och mer tillförlitliga resultat. 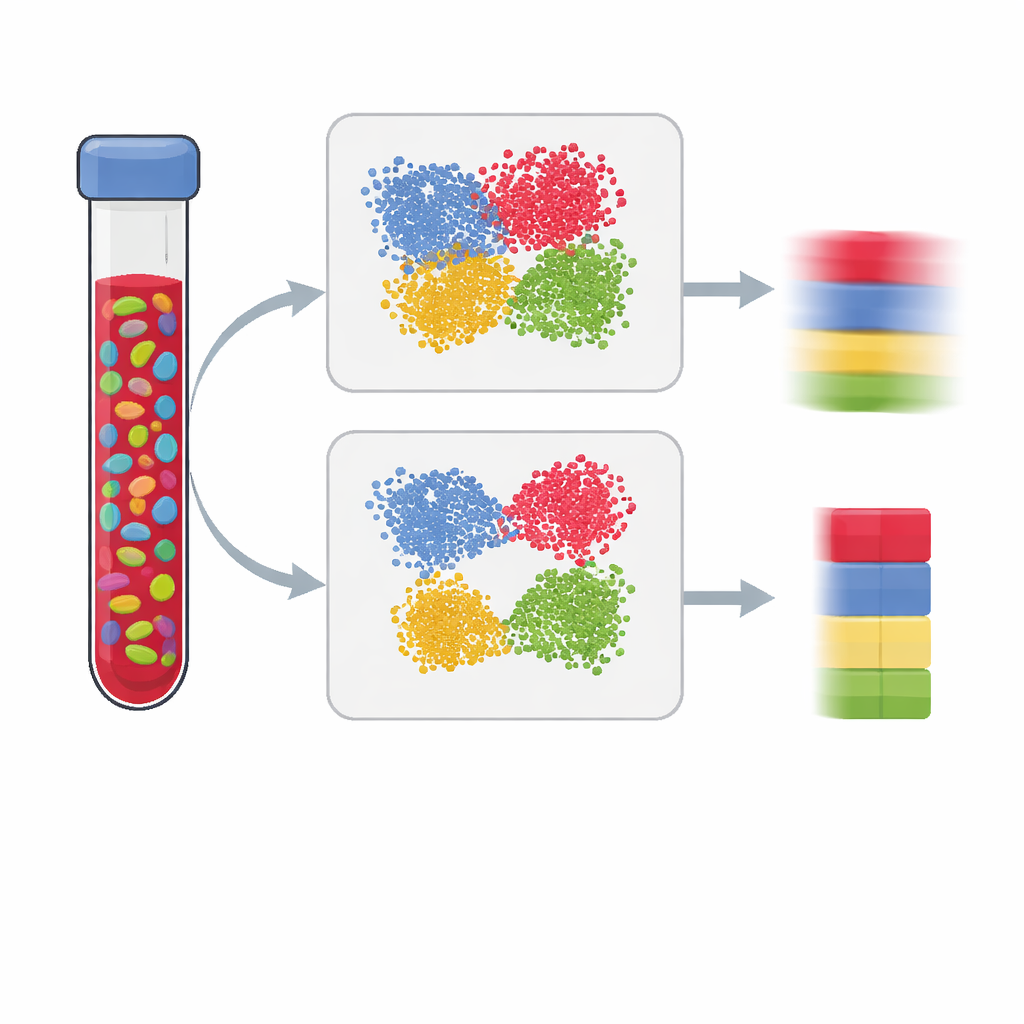
Varför cellblandning spelar roll
Epigenomvida associationsstudier söker efter skillnader i DNA-metylering — tillsats av små kemiska grupper som hjälper till att reglera genaktivitet — mellan personer med och utan ett visst drag, till exempel en sjukdom. Eftersom metyleringsmönster skiljer sig kraftigt mellan celltyper kan mätningar i bulkblod bli missvisande: en förskjutning från en immuncelltyp till en annan kan efterlikna en sjukdomseffekt även om inget förändrats inom varje celltyp. För att korrigera detta uppskattar forskare andelarna av stora celltyper (såsom T‑celler, B‑celler och naturliga mördarceller) med hjälp av en referenspanel byggd från renade celler eller enskilda celler. Panelens kvalitet avgör i stor utsträckning hur väl vi kan ”avblanda” ett prov och därmed hur tillförlitliga studiens slutsatser blir.
Från enkel statistik till smartare markörer
Traditionellt valde forskare DNA‑positioner för dessa paneler med standardstatistiska tester. Man sökte positioner där en celltyp skilde sig signifikant från alla andra och rankade dem med en t‑statistik. På senare tid har optimerings- och maskininlärningsmetoder som IDOL, Elastic Net och Random Forests använts för att förfina dessa val. Den nya studien visar att dessa angreppssätt ofta prioriterar markörer med små verkliga skillnader mellan celltyper, särskilt när man har bara ett fåtal renade prov. Sådana markörer med ”liten effektstorlek” kan se övertygande ut i träningdata men misslyckas i nya dataset och därigenom subtilt försämra noggrannheten i celltypuppskattningarna.
Att hitta tydliga luckor mellan celltyper
Författarna föreslår ett mer direkt sätt att bedöma hur användbar en markör är: en ”gap‑specificitetspoäng”. Istället för att bara fokusera på statistisk signifikans mäter denna poäng hur rent en DNA‑position separerar en celltyp från alla andra genom att titta på gapet mellan det högsta värdet i målcellstypen och det lägsta värdet i alla andra celler (eller omvänt för låga värden). Markörer med stora positiva gap är både specifika och robusta. Med befintliga data för immunceller visade forskarna att rangordning av markörer efter denna poäng ger DNA‑positioner med mycket större skillnader mellan celltyper än den traditionella metoden. Paneler byggda av dessa gap‑baserade markörer gav mer korrekta uppskattningar av cellfraktioner över många immunsubset, särskilt för svårare att separera populationer som minnes CD4‑T‑celler. 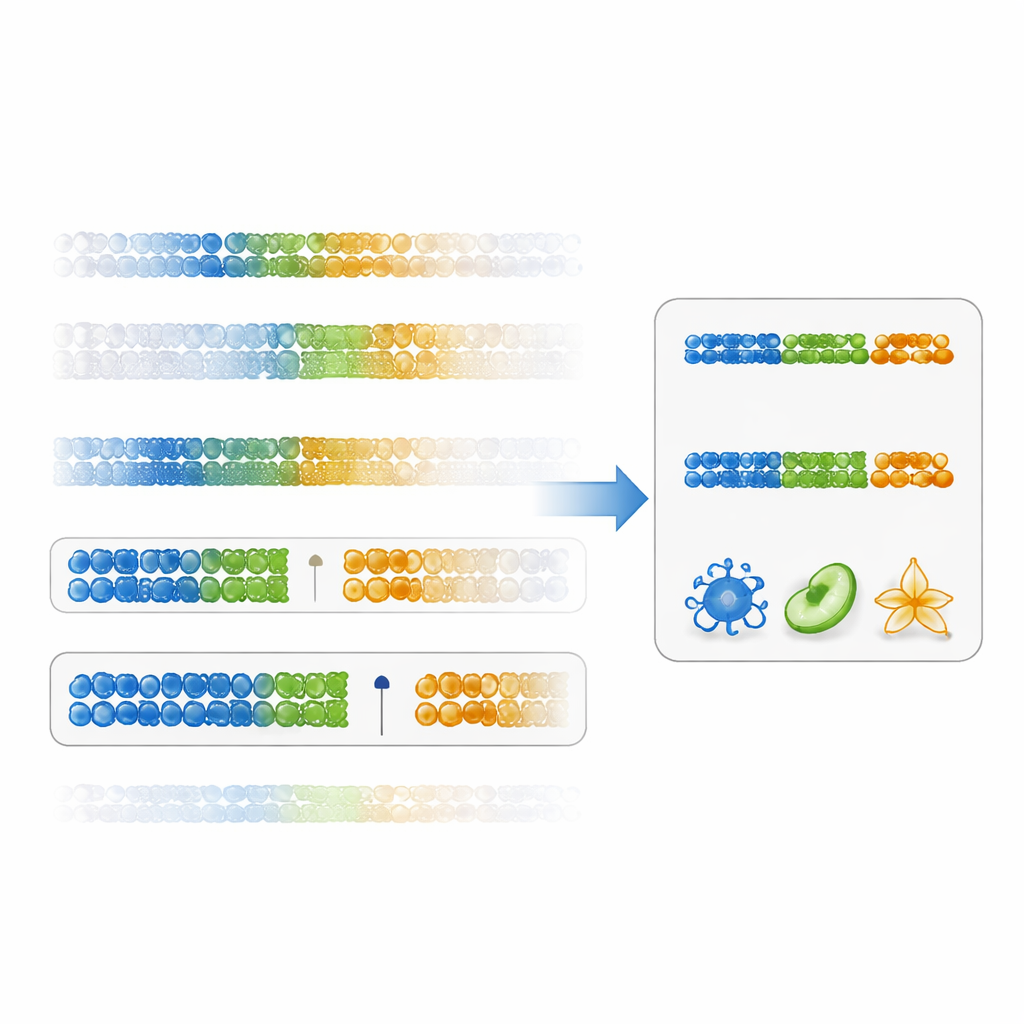
Varför låga effektstorlekar och överanpassning skadar
Gruppen testade även om avancerade optimeringsverktyg eller maskininlärningsmodeller kunde förbättra deras gap‑baserade angreppssätt. Istället fann de motsatsen. Metoder som IDOL, Elastic Net och Random Forest tenderade att välja funktioner med mindre effektstorlek och presterade sämre när de utvärderades på oberoende blandningar eller på verkliga blodprover med kända cellantal. Detta tyder på att komplexa modeller med bara några tiotal träningsprov överanpassar dataegenskaper i stället för att fånga generella mönster. I kontrast förbättrade paneler byggda enbart av starkt hypometylerade markörer med höga gap‑poäng inte bara dekonvolutionsnoggrannheten utan fångade också bättre kända biologiska trender, såsom åldersrelaterad ökning av naturliga mördarceller.
Tydligare sjukdomssignaler tack vare bättre paneler
För att visa hur dessa förbättringar påverkar praktiken analyserade författarna om stora studier av schizofreni och typ 1‑diabetes. Med deras optimerade referenspaneler förändrades de uppskattade cellfraktionerna endast något, men dessa små skift stärkte efterföljande fynd. Sjukdomsassocierade metyleringsförändringar blev mer fördjupade i vägar som redan implicerats i inflammation och autoimmunitet, och specifika gener kopplade till immunsignalering framträdde tydligare. Med andra ord minskade bättre markörval brus och gjorde den biologiska berättelsen mer sammanhängande.
Vad detta betyder för framtida studier
För icke‑specialister är huvudbudskapet att inte alla statistiskt signifikanta signaler är lika användbara. När man försöker reda ut blandade vävnader är det viktigaste hur tydligt en markör särskiljer en celltyp från en annan, inte bara hur imponerande dess P‑värde ser ut. Genom att prioritera DNA‑positioner med stora, rena gap mellan celltyper — särskilt sådana som är unikt ometylerade i en given cell — kan forskare bygga mer tillförlitliga referenspaneler även från små dataset. Författarna har lagt till verktyg för att konstruera sådana paneler i mjukvaran EpiDISH, vilket hjälper framtida studier att dra mer exakta och biologiskt meningsfulla slutsatser från DNA‑metyleringsdata.
Citering: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Nyckelord: DNA-metylering, cell-typ dekonvolution, epigenomik, immunceller, referenspaneler