Clear Sky Science · nl
Richtlijnen voor het optimaliseren van referentiepanelen voor DNA-methylering bij celtype-deconvolutie
Inzicht in gemengde weefsels
Moderne onderzoeken naar gezondheid en ziekte meten vaak chemische labels op ons DNA om te begrijpen hoe omgeving en levensstijl sporen achterlaten in onze genen. De meeste tests gebeuren echter op gemengde weefsels zoals bloed, die uit veel verschillende celtypen bestaan. Als we niet kunnen bepalen hoeveel van elk celtype aanwezig is, kunnen we een verandering in de cel-samenstelling verwarren met een echte ziektesignaal. Dit artikel beschrijft hoe je betere “referentiepanelen” kunt opbouwen die wetenschappers in staat stellen om celmengsels nauwkeurig te schatten op basis van DNA-methyleringsdata, wat leidt tot helderdere en betrouwbaardere resultaten. 
Waarom de celverhouding ertoe doet
Epigenoom-brede associatiestudies zoeken naar verschillen in DNA-methylering—de toevoeging van kleine chemische labels die genactiviteit helpen reguleren—tussen mensen met en zonder een eigenschap, zoals een ziekte. Omdat methyleringspatronen sterk verschillen tussen celtypen, kan meten in bulkbloed misleidend zijn: een verschuiving van het ene immuunceltype naar het andere kan een ziekte-effect nadoen, zelfs als binnen elk celtype niets veranderde. Om dit te corrigeren schatten onderzoekers de fracties van de belangrijkste celtypen (zoals T-cellen, B-cellen en natural killer-cellen) met behulp van een referentiepanel dat is opgebouwd uit gezuiverde cellen of enkele cellen. De kwaliteit van dat paneel bepaalt grotendeels hoe goed we een monster kunnen “ontmengen” en daarmee hoe betrouwbaar de conclusies van een studie zijn.
Van eenvoudige statistiek naar slimmer gekozen markers
Traditioneel kozen wetenschappers DNA-plaatsen voor deze panelen met behulp van standaard statistische testen. Ze zochten posities waar één celtype significant verschilde van alle andere en rangschikten die op basis van een t-statistiek. Recente optimalisatie- en machine-learningmethoden zoals IDOL, Elastic Net en Random Forests worden gebruikt om deze keuzes te verfijnen. De nieuwe studie toont aan dat deze benaderingen vaak markers prioriteren met kleine werkelijke verschillen tussen celtypen, vooral wanneer slechts een handvol gezuiverde monsters beschikbaar is. Dergelijke markers met "lage effectgrootte" kunnen overtuigend lijken in trainingsdata maar falen in nieuwe datasets, waardoor de nauwkeurigheid van celtypeschattingen subtiel verslechtert.
Grote verschillen tussen celtypen vinden
De auteurs stellen een directere manier voor om de bruikbaarheid van een marker te beoordelen: een "gap-specificity score" (kloofscorespecificiteit). In plaats van alleen naar statistische significantie te kijken, meet deze score hoe schoon een DNA-locatie één celtype scheidt van alle anderen, door te kijken naar de kloof tussen de hoogste waarde in het doelceltype en de laagste waarde in alle andere cellen (of omgekeerd voor lage waarden). Markers met grote positieve kloven zijn zowel specifiek als robuust. Met bestaande immuuncelgegevens lieten de onderzoekers zien dat het rangschikken van markers op basis van deze score DNA-plaatsen oplevert met veel grotere verschillen tussen celtypen dan de traditionele methode. Panelen opgebouwd uit deze kloof-gebaseerde markers leverden nauwkeurigere schattingen van celfracties op voor vele immuunsubsets, met name voor moeilijker te onderscheiden populaties zoals geheugen CD4-T-cellen. 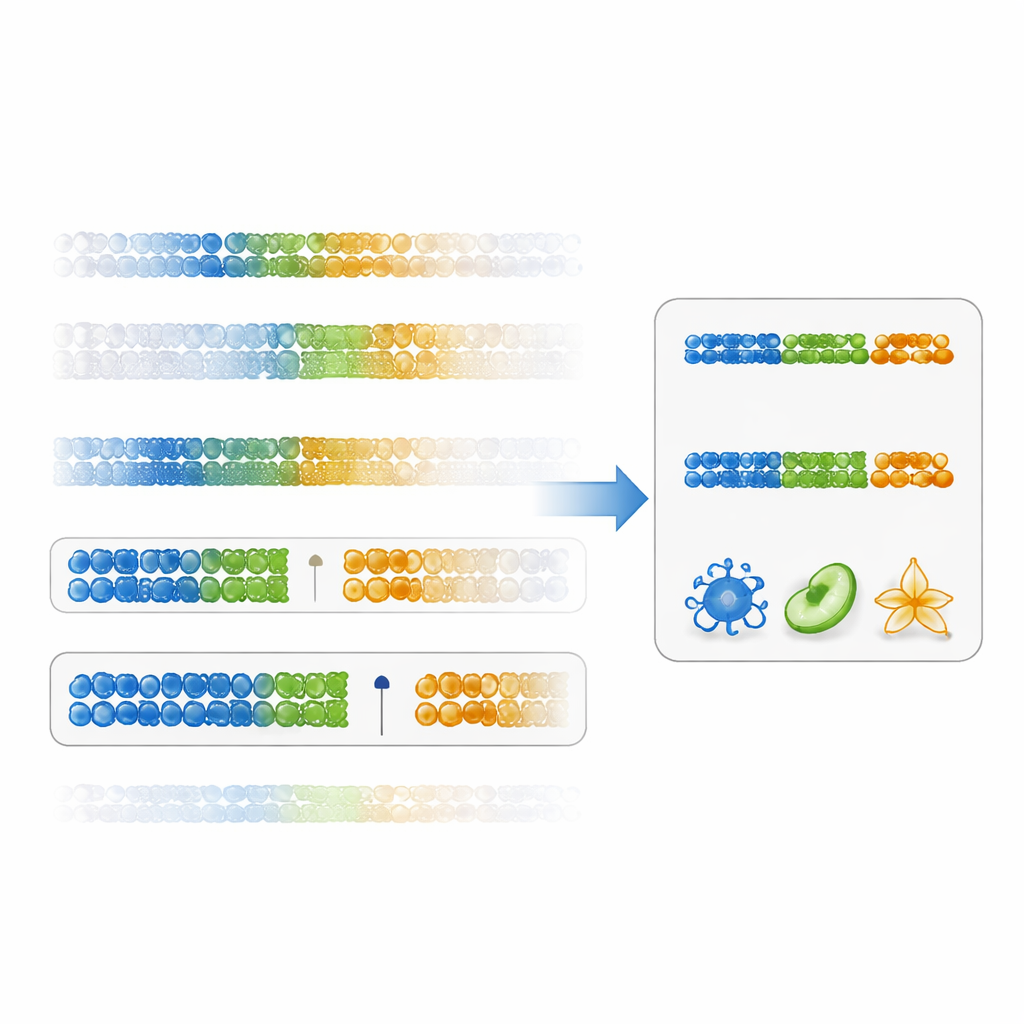
Waarom lage scores en overfitting schadelijk zijn
Het team testte ook of geavanceerde optimalisatie-instrumenten of machine-learningmodellen beter konden presteren dan hun kloof-gebaseerde aanpak. In plaats daarvan vonden ze het omgekeerde. Methoden zoals IDOL, Elastic Net en Random Forest selecteerden vaak kenmerken met kleinere effectgroottes en presteerden slechter bij evaluatie op onafhankelijke mengsels of op echt bloed met bekende celgetallen. Dit suggereert dat, met slechts enkele tientallen trainingsmonsters, complexe modellen de eigenaardigheden van de data overfitten in plaats van algemene patronen te leren. Daarentegen verbeterden panelen die uitsluitend bestonden uit sterk hypomethyleerde markers met hoge kloofscores niet alleen de deconvolutie-nauwkeurigheid, maar vingen ze ook beter bekende biologische trends, zoals de leeftijdsgerelateerde toename van natural killer-cellen.
Helderdere ziektesignalen door betere panelen
Om te laten zien hoe deze verbeteringen in de praktijk uitpakken, heranalyseerden de auteurs grote studies naar schizofrenie en type 1-diabetes. Het gebruik van hun geoptimaliseerde referentiepanelen veranderde de geschatte celfracties slechts licht, maar die kleine verschuivingen verscherpten de downstream-resultaten. Ziekte-geassocieerde methyleringsveranderingen werden sterker verrijkt voor paden die al met ontsteking en auto-immuniteit in verband worden gebracht, en specifieke genen die betrokken zijn bij immuunsignalering kwamen duidelijker naar voren. Met andere woorden: betere markerselectie verminderde ruis en maakte het biologische verhaal coherenter.
Wat dit betekent voor toekomstige studies
Voor niet-specialisten is de kernboodschap dat niet alle statistisch significante signalen even nuttig zijn. Bij het ontwarren van gemengde weefsels is het belangrijkste hoe duidelijk een marker één celtype van een ander onderscheidt, en niet alleen hoe indrukwekkend de p-waarde lijkt. Door de voorkeur te geven aan DNA-plaatsen met grote, schone kloven tussen celtypen—vooral diegenen die uniek unmethyleerd zijn in een bepaald celtype—kunnen onderzoekers betrouwbare referentiepanelen bouwen, zelfs uit kleine datasets. De auteurs hebben hulpmiddelen voor het construeren van dergelijke panelen toegevoegd aan de EpiDISH-software, wat toekomstige studies helpt om nauwkeurigere en biologisch betekenisvollere conclusies te trekken uit DNA-methyleringsdata.
Bronvermelding: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Trefwoorden: DNA-methylering, celtype-deconvolutie, epigenomics, immuuncellen, referentiepanelen