Clear Sky Science · it
Linee guida per ottimizzare i pannelli di riferimento della metilazione del DNA per la deconvoluzione dei tipi cellulari
Uno sguardo all’interno dei tessuti misti
Gli studi moderni sulla salute e la malattia spesso misurano modifiche chimiche sul nostro DNA, per capire come ambiente e stile di vita lascino tracce sui geni. Ma la maggior parte dei test viene eseguita su tessuti misti come il sangue, che contengono molti tipi cellulari. Se non sappiamo quanto è presente ciascun tipo cellulare, potremmo scambiare una variazione nella composizione cellulare per un vero segnale di malattia. Questo articolo spiega come costruire «pannelli di riferimento» migliori che permettono agli scienziati di stimare con precisione le miscele cellulari dai dati di metilazione del DNA, portando a risultati più chiari e affidabili. 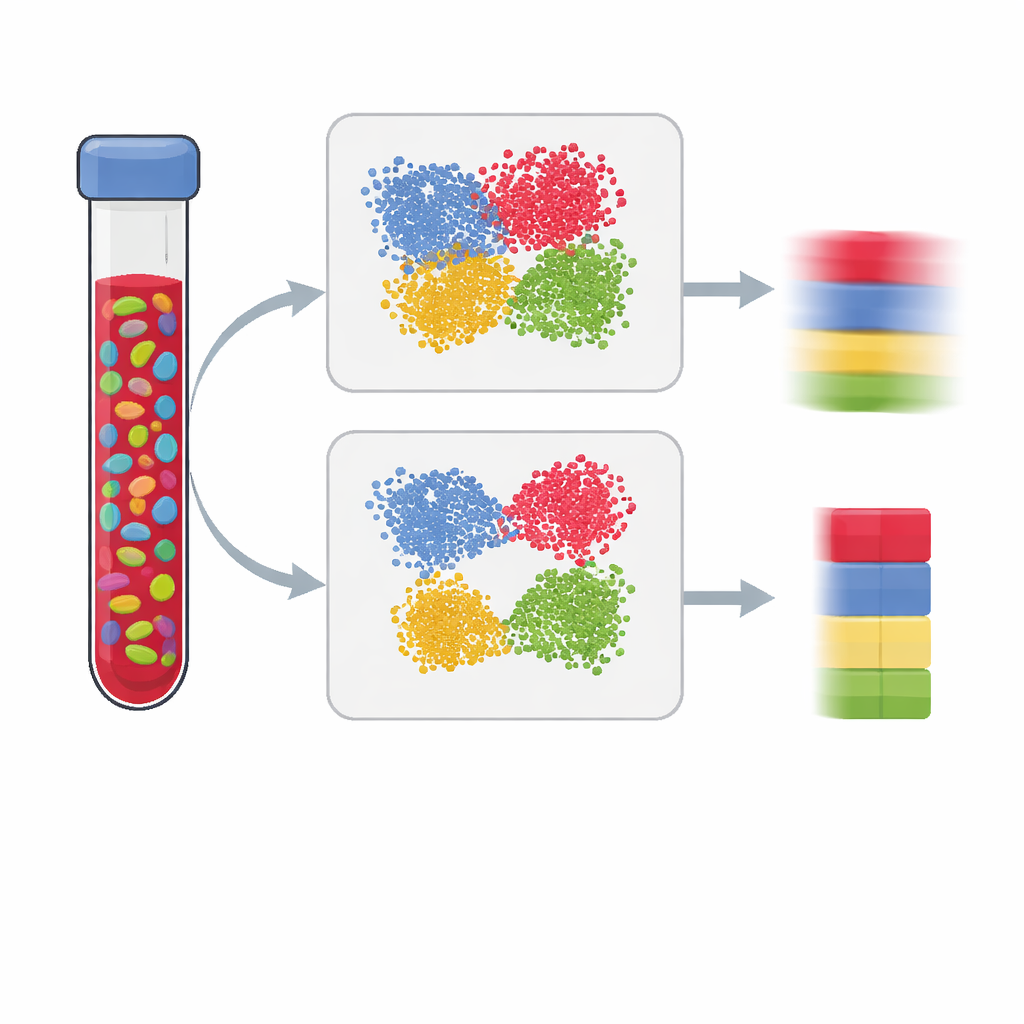
Perché la composizione cellulare conta
Gli studi di associazione epigenome-wide cercano differenze nella metilazione del DNA — l’aggiunta di piccoli segni chimici che aiutano a controllare l’attività genica — tra persone con e senza un tratto, come una malattia. Poiché i pattern di metilazione variano molto tra i diversi tipi cellulari, misurarli in campioni bulk può essere fuorviante: uno spostamento da un tipo di cellula immunitaria a un altro può imitare un effetto di malattia anche se nulla è cambiato all’interno di ciascun tipo cellulare. Per correggere questo, i ricercatori stimano le frazioni dei principali tipi cellulari (come cellule T, cellule B e cellule natural killer) usando un pannello di riferimento costruito a partire da cellule purificate o cellule singole. La qualità di quel pannello determina in gran parte quanto bene possiamo «scomporre» un campione e, di conseguenza, quanto sono affidabili le conclusioni dello studio.
Dalla statistica semplice a marcatori più intelligenti
Tradizionalmente, gli scienziati selezionavano siti del DNA per questi pannelli usando test statistici standard. Cercavano posizioni in cui un tipo cellulare differiva significativamente da tutti gli altri e le classificavano per statistica t. Più recentemente, metodi di ottimizzazione e apprendimento automatico come IDOL, Elastic Net e Random Forest sono stati impiegati per perfezionare queste scelte. Il nuovo studio mostra che questi approcci spesso privilegiano marcatori con piccole differenze effettive tra i tipi cellulari, soprattutto quando sono disponibili solo poche decine di campioni purificati. Tali marcatori a «bassa dimensione d’effetto» possono sembrare convincenti nei dati di addestramento ma fallire in nuovi dataset, degradando sottilmente la precisione delle stime delle frazioni cellulari.
Trovare gap netti tra i tipi cellulari
Gli autori propongono un modo più diretto per valutare l’utilità di un marcatore: un «punteggio di specificità del gap». Invece di concentrarsi solo sulla significatività statistica, questo punteggio misura quanto nettamente un sito del DNA separa un tipo cellulare da tutti gli altri, osservando il divario tra il valore più alto nel tipo target e il valore più basso in tutte le altre cellule (o viceversa per valori bassi). I marcatori con gap positivi ampi sono sia specifici sia robusti. Usando dati esistenti su cellule immunitarie, i ricercatori hanno mostrato che classificare i marcatori con questo punteggio produce siti del DNA con differenze tra tipi cellulari molto più grandi rispetto al metodo tradizionale. I pannelli costruiti da questi marcatori basati sul gap hanno fornito stime delle frazioni cellulari più accurate attraverso molti sottogruppi immunitari, in particolare per popolazioni più difficili da risolvere come le cellule T CD4 della memoria. 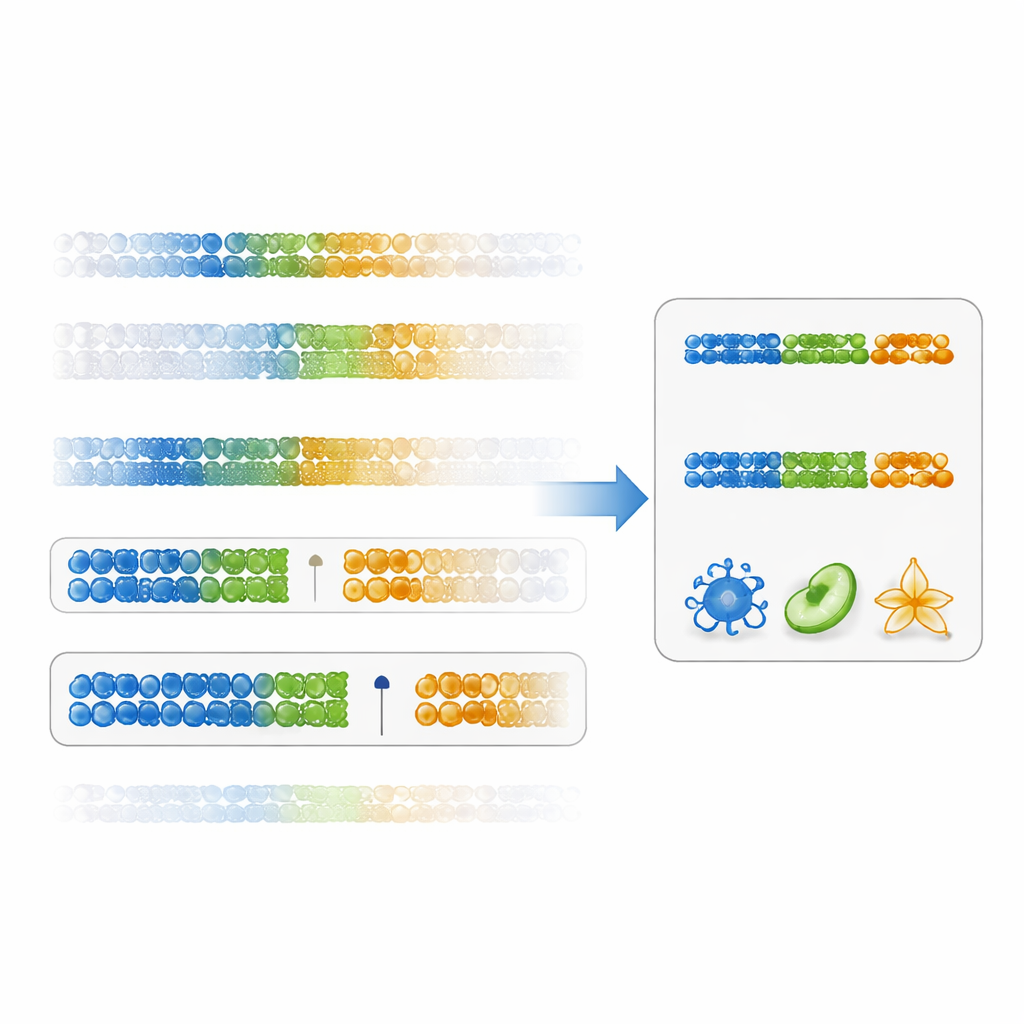
Perché i marcatori deboli e l’overfitting danneggiano
Il team ha anche testato se strumenti di ottimizzazione avanzati o modelli di machine learning potessero migliorare l’approccio basato sul gap. Al contrario, hanno riscontrato l’effetto opposto. Metodi come IDOL, Elastic Net e Random Forest tendevano a selezionare caratteristiche con dimensioni d’effetto più piccole e prestavano peggio quando valutati su miscele indipendenti o su campioni di sangue reali con conte cellulari noti. Ciò suggerisce che, con solo poche decine di campioni di addestramento, i modelli complessi overfittano peculiarità dei dati invece di catturare schemi generali. Al contrario, pannelli costruiti esclusivamente da marcatori fortemente ipometilati con punteggi di gap elevati non solo miglioravano l’accuratezza della deconvoluzione, ma coglievano anche meglio trend biologici noti, come l’aumento correlato all’età delle cellule natural killer.
Segnali di malattia più chiari grazie a pannelli migliori
Per vedere come questi miglioramenti si traducono nella pratica, gli autori hanno rianalizzato grandi studi su schizofrenia e diabete di tipo 1. L’uso dei loro pannelli di riferimento ottimizzati ha modificato le frazioni cellulari stimate solo leggermente, ma quei piccoli spostamenti hanno reso più nitidi i risultati successivi. Le modifiche di metilazione associate alla malattia sono risultate più arricchite per vie già implicate nell’infiammazione e nell’autoimmunità, e geni specifici legati alla segnalazione immunitaria sono emersi più chiaramente. In altre parole, una selezione migliore dei marcatori ha ridotto il rumore e reso la narrazione biologica più coerente.
Cosa significa per gli studi futuri
Per i non specialisti, il messaggio chiave è che non tutti i segnali statisticamente significativi sono ugualmente utili. Quando si cerca di districare tessuti misti, ciò che conta di più è quanto chiaramente un marcatore distingue un tipo cellulare da un altro, non solo quanto impressionante sembra il suo valore p. Favorendo siti del DNA con gap ampi e netti tra i tipi cellulari — in particolare quelli unicamente ipometilati in un dato tipo cellulare — i ricercatori possono costruire pannelli di riferimento più affidabili anche da dataset piccoli. Gli autori hanno aggiunto strumenti per costruire tali pannelli al software EpiDISH, aiutando gli studi futuri a ricavare conclusioni più accurate e biologicamente significative dai dati di metilazione del DNA.
Citazione: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Parole chiave: Metilazione del DNA, deconvoluzione dei tipi cellulari, epigenomica, cellule immunitarie, pannelli di riferimento