Clear Sky Science · es
Directrices para optimizar paneles de referencia de metilación del ADN para la deconvolución de tipos celulares
Mirando dentro de tejidos mixtos
Los estudios modernos sobre salud y enfermedad a menudo miden marcas químicas en nuestro ADN con la esperanza de ver cómo el entorno y el estilo de vida dejan rastros en nuestros genes. Pero la mayoría de las pruebas se realizan en tejidos mixtos como la sangre, que contienen muchos tipos celulares. Si no podemos saber cuánto de cada tipo celular está presente, podemos confundir un cambio en la composición celular con una señal verdadera de enfermedad. Este artículo explica cómo construir mejores “paneles de referencia” que permiten a los científicos estimar con precisión las mezclas celulares a partir de datos de metilación del ADN, conduciendo a resultados más claros y fiables. 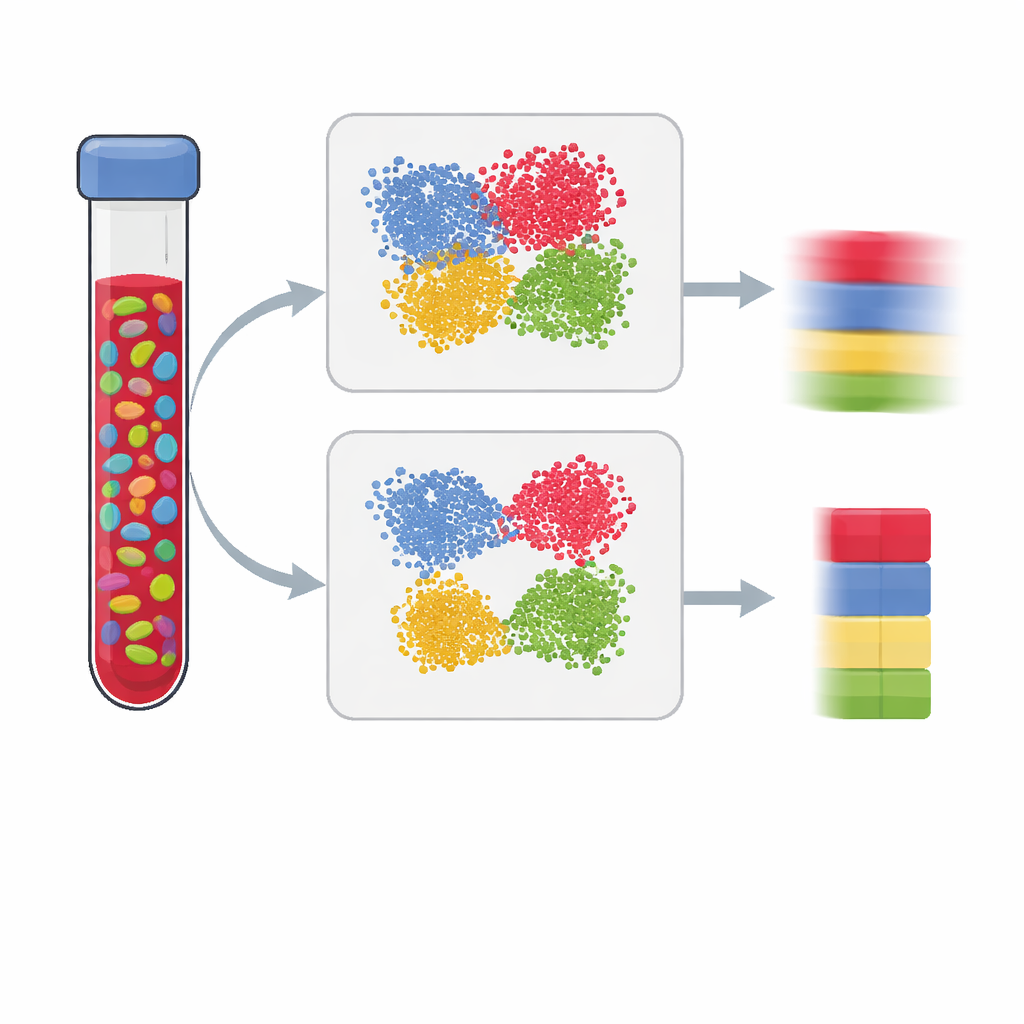
Por qué importa la mezcla celular
Los estudios de asociación epigenómica buscan diferencias en la metilación del ADN —la adición de pequeñas marcas químicas que ayudan a regular la actividad génica— entre personas con y sin un rasgo, como una enfermedad. Dado que los patrones de metilación varían mucho entre tipos celulares, medirlos en sangre total puede ser engañoso: un cambio de un tipo de célula inmunitaria a otro puede imitar un efecto de enfermedad aunque no haya cambios dentro de cada tipo celular. Para corregir esto, los investigadores estiman las fracciones de los principales tipos celulares (por ejemplo, células T, células B y células NK) usando un panel de referencia construido a partir de células purificadas o de células individuales. La calidad de ese panel determina en gran medida cuán bien podemos “desmezclar” una muestra y, por ende, cuán fiables son las conclusiones del estudio.
De la estadística simple a marcadores más inteligentes
Tradicionalmente, los científicos seleccionaban sitios de ADN para estos paneles usando pruebas estadísticas estándar. Buscaban posiciones donde un tipo celular difiriera significativamente de todos los demás y las ordenaban por un estadístico t. Más recientemente, métodos de optimización y aprendizaje automático como IDOL, Elastic Net y Random Forests se han utilizado para refinar estas elecciones. El estudio nuevo muestra que estos enfoques con frecuencia priorizan marcadores con diferencias pequeñas en el mundo real entre tipos celulares, especialmente cuando solo hay unas pocas muestras purificadas disponibles. Tales marcadores de “bajo tamaño del efecto” pueden parecer convincentes en los datos de entrenamiento pero fallar en conjuntos de datos nuevos, degradando sutilmente la precisión de las estimaciones de los tipos celulares.
Encontrar brechas claras entre tipos celulares
Los autores proponen una forma más directa de juzgar la utilidad de un marcador: una “puntuación de especificidad por brecha”. En lugar de centrarse únicamente en la significación estadística, esta puntuación mide qué tan nítidamente un sitio de ADN separa un tipo celular de todos los demás, mirando la brecha entre el valor más alto en la célula objetivo y el valor más bajo en todas las demás células (o al revés para valores bajos). Los marcadores con grandes brechas positivas son tanto específicos como robustos. Usando datos existentes de células inmunitarias, los investigadores mostraron que ordenar marcadores por esta puntuación produce sitios de ADN con diferencias mucho mayores entre tipos celulares que el método tradicional. Los paneles construidos a partir de estos marcadores basados en brechas ofrecieron estimaciones de fracción celular más precisas en muchos subconjuntos inmunitarios, particularmente para poblaciones más difíciles de resolver como las células T CD4 de memoria. 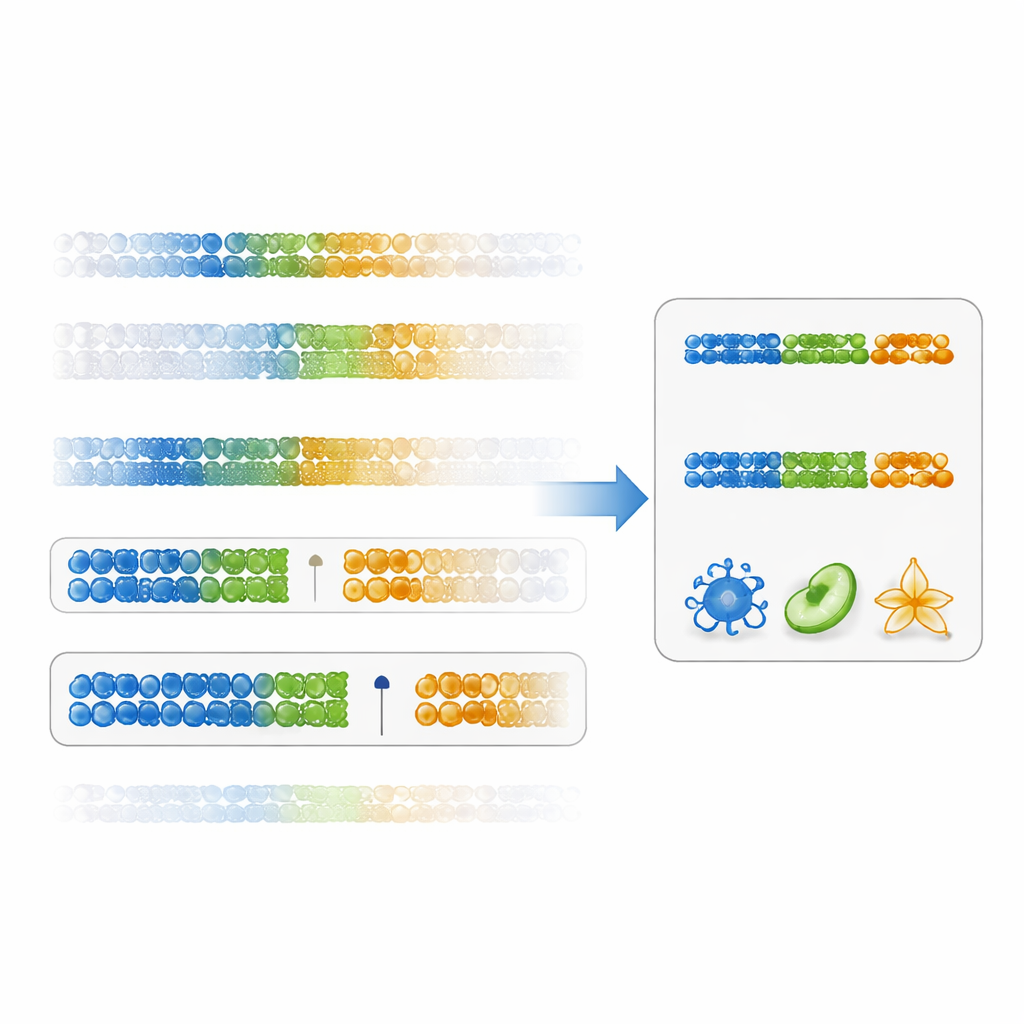
Por qué las marcas pequeñas y el sobreajuste perjudican
El equipo también probó si herramientas de optimización avanzadas o modelos de aprendizaje automático podían mejorar su enfoque basado en brechas. En cambio, encontraron lo opuesto. Métodos como IDOL, Elastic Net y Random Forest tendieron a seleccionar características con tamaños de efecto más pequeños y rindieron peor cuando se evaluaron en mezclas independientes o en muestras de sangre reales con recuentos celulares conocidos. Esto sugiere que, con solo unas pocas docenas de muestras de entrenamiento, los modelos complejos sobreajustan particularidades de los datos en lugar de capturar patrones generales. En contraste, los paneles construidos únicamente a partir de marcadores fuertemente hipometilados con altas puntuaciones de brecha no solo mejoraron la exactitud de la deconvolución sino que también captaron mejor tendencias biológicas conocidas, como el aumento relacionado con la edad en las células NK.
Señales de enfermedad más claras gracias a mejores paneles
Para ver cómo se traducen estas mejoras en la práctica, los autores reanalizaron grandes estudios sobre esquizofrenia y diabetes tipo 1. El uso de sus paneles de referencia optimizados cambió las fracciones celulares estimadas solo ligeramente, pero esos pequeños desplazamientos agudizaron los hallazgos posteriores. Los cambios en la metilación asociados a la enfermedad se enriquecieron más en vías ya implicadas en inflamación y autoinmunidad, y genes específicos relacionados con la señalización inmune emergieron con mayor claridad. En otras palabras, una mejor selección de marcadores redujo el ruido y hizo que la historia biológica fuera más coherente.
Qué significa esto para estudios futuros
Para no especialistas, el mensaje clave es que no todas las señales estadísticamente significativas son igualmente útiles. Al intentar desenredar tejidos mixtos, lo que más importa es cuán claramente un marcador distingue un tipo celular de otro, no solo lo impresionante que parezca su valor P. Al favorecer sitios de ADN con brechas grandes y limpias entre tipos celulares —especialmente aquellos que están desmetilados de forma única en una célula dada— los investigadores pueden construir paneles de referencia más fiables incluso a partir de conjuntos de datos pequeños. Los autores han añadido herramientas para construir tales paneles al software EpiDISH, ayudando a que estudios futuros obtengan conclusiones más precisas y biológicamente significativas a partir de datos de metilación del ADN.
Cita: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Palabras clave: Metilación del ADN, Deconvolución de tipos celulares, Epigenómica, Células inmunitarias, Paneles de referencia