Clear Sky Science · en
Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution
Peering Inside Mixed Tissues
Modern studies of health and disease often measure chemical tags on our DNA, hoping to see how environment and lifestyle leave marks on our genes. But most tests are run on mixed tissues like blood, which contain many kinds of cells. If we cannot tell how much of each cell type is present, we may mistake a change in cell mixture for a true disease signal. This paper explains how to build better “reference panels” that let scientists accurately estimate cell mixtures from DNA methylation data, leading to clearer, more trustworthy results. 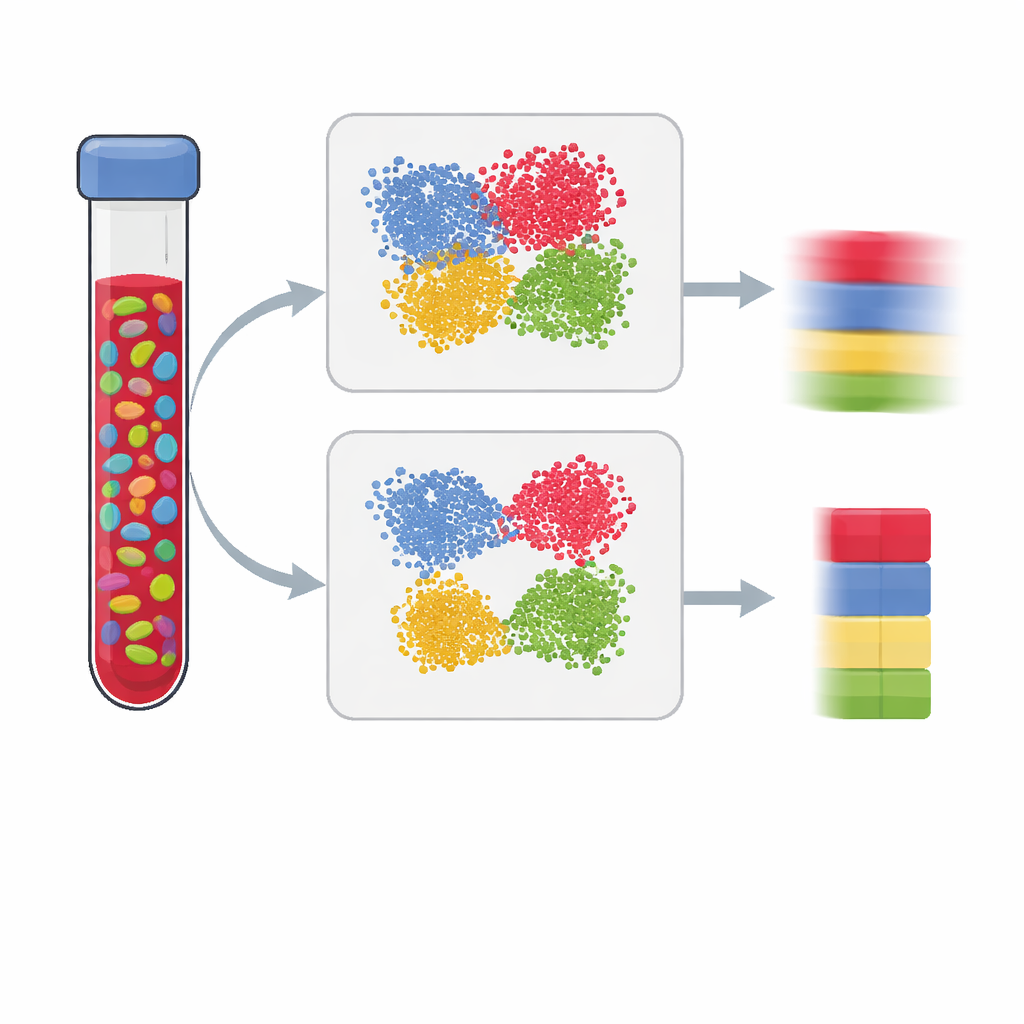
Why Cell Mix Matters
Epigenome-wide association studies look for differences in DNA methylation—the addition of small chemical tags that help control gene activity—between people with and without a trait, such as a disease. Because methylation patterns are very different from one cell type to another, measuring them in bulk blood can be misleading: a shift from one immune cell type to another can mimic a disease effect even if nothing changed within each cell type. To correct this, researchers estimate the fractions of major cell types (such as T cells, B cells, and natural killer cells) using a reference panel built from purified cells or single cells. The quality of that panel largely determines how well we can “unmix” a sample and, in turn, how reliable the study’s conclusions are.
From Simple Statistics to Smarter Markers
Traditionally, scientists picked DNA sites for these panels using standard statistical tests. They searched for positions where one cell type differed significantly from all others and ranked them by a t-statistic. More recently, optimization and machine-learning methods such as IDOL, Elastic Net, and Random Forests have been used to refine these choices. The new study shows that these approaches often prioritize markers with small real-world differences between cell types, especially when only a handful of purified samples are available. Such “low-effect-size” markers may look convincing in the training data but fail in new datasets, subtly degrading the accuracy of cell-type estimates.
Finding Clear Gaps Between Cell Types
The authors propose a more direct way to judge how useful a marker is: a “gap specificity score.” Instead of focusing only on statistical significance, this score measures how cleanly a DNA site separates one cell type from all others, by looking at the gap between the highest value in the target cell and the lowest value in all other cells (or the reverse for low values). Markers with large positive gaps are both specific and robust. Using existing immune-cell data, the researchers showed that ranking markers by this score yields DNA sites with much larger differences between cell types than the traditional method. Panels built from these gap-based markers produced more accurate cell-fraction estimates across many immune subsets, particularly for harder-to-resolve populations like memory CD4 T cells. 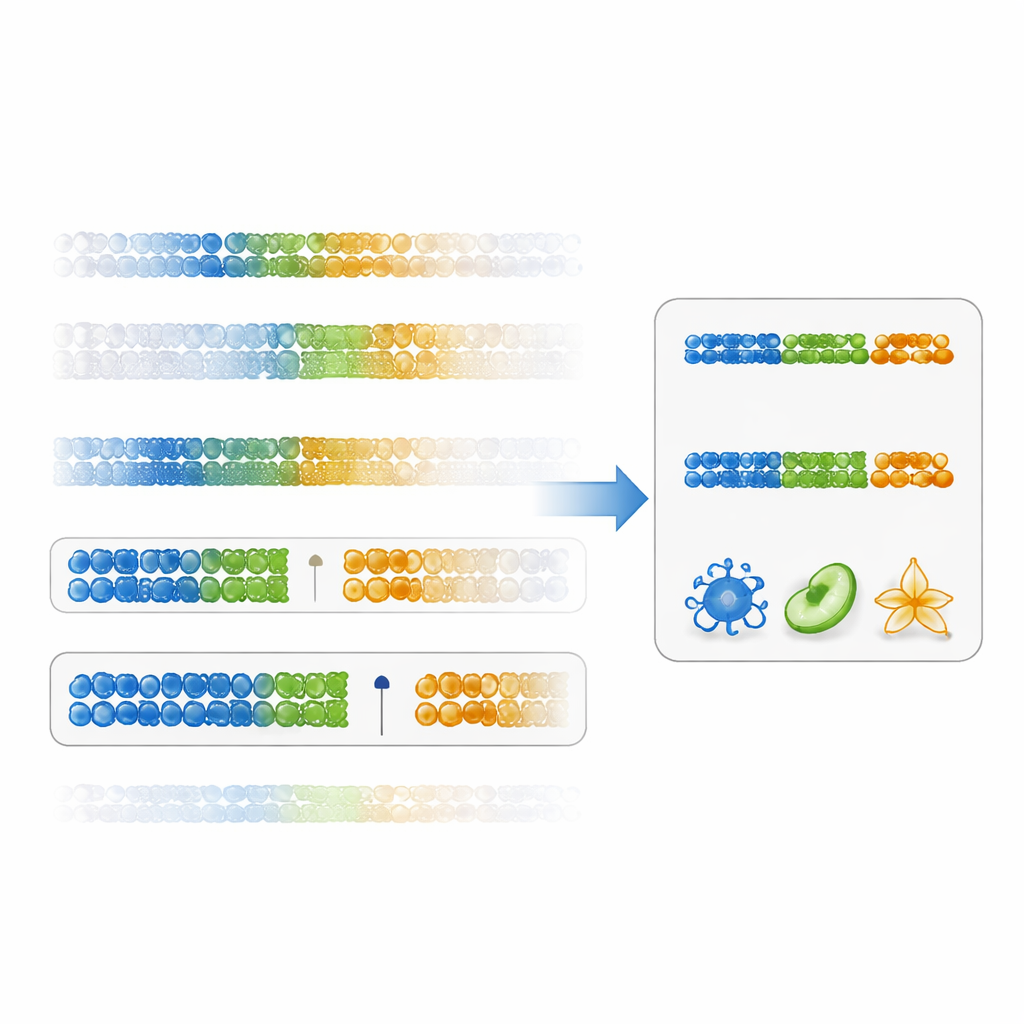
Why Low Marks and Overfitting Hurt
The team also tested whether advanced optimization tools or machine-learning models could improve on their gap-based approach. Instead, they found the opposite. Methods like IDOL, Elastic Net, and Random Forest tended to select features with smaller effect sizes and performed worse when evaluated on independent mixtures or on real blood samples with known cell counts. This suggests that, with only a few dozen training samples, complex models overfit quirks of the data rather than capturing general patterns. In contrast, panels built solely from strongly hypomethylated markers with high gap scores not only improved deconvolution accuracy but also better captured known biological trends, such as the age-related rise in natural killer cells.
Clearer Disease Signals from Better Panels
To see how these improvements play out in practice, the authors reanalyzed large studies of schizophrenia and type 1 diabetes. Using their optimized reference panels changed the estimated cell fractions only slightly, but those small shifts sharpened downstream findings. Disease-associated methylation changes became more enriched for pathways already implicated in inflammation and autoimmunity, and specific genes linked to immune signaling emerged more clearly. In other words, better marker selection reduced noise and made the biological story more coherent.
What This Means for Future Studies
For non-specialists, the key message is that not all statistically significant signals are equally useful. When trying to untangle mixed tissues, what matters most is how clearly a marker distinguishes one cell type from another, not just how impressive its P-value looks. By favoring DNA sites with large, clean gaps between cell types—especially those that are uniquely unmethylated in a given cell—researchers can build more reliable reference panels even from small datasets. The authors have added tools for constructing such panels to the EpiDISH software, helping future studies draw more accurate and biologically meaningful conclusions from DNA methylation data.
Citation: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Keywords: DNA methylation, cell-type deconvolution, epigenomics, immune cells, reference panels