Clear Sky Science · pl
Wytyczne dotyczące optymalizacji paneli referencyjnych metylacji DNA do dekonwolucji typów komórek
Zaglądając w mieszane tkanki
Współczesne badania nad zdrowiem i chorobami często mierzą chemiczne znaczniki na naszym DNA, aby zobaczyć, jak środowisko i styl życia pozostawiają ślady na genach. Jednak większość analiz przeprowadza się na mieszanych tkankach, takich jak krew, które zawierają wiele rodzajów komórek. Jeśli nie potrafimy określić udziału poszczególnych typów komórek, możemy pomylić zmianę składu komórkowego ze skutecznym sygnałem chorobowym. W artykule tym opisano, jak budować lepsze „panele referencyjne”, które pozwalają naukowcom dokładnie oszacować mieszankę komórek na podstawie danych o metylacji DNA, co prowadzi do jaśniejszych i bardziej wiarygodnych wyników. 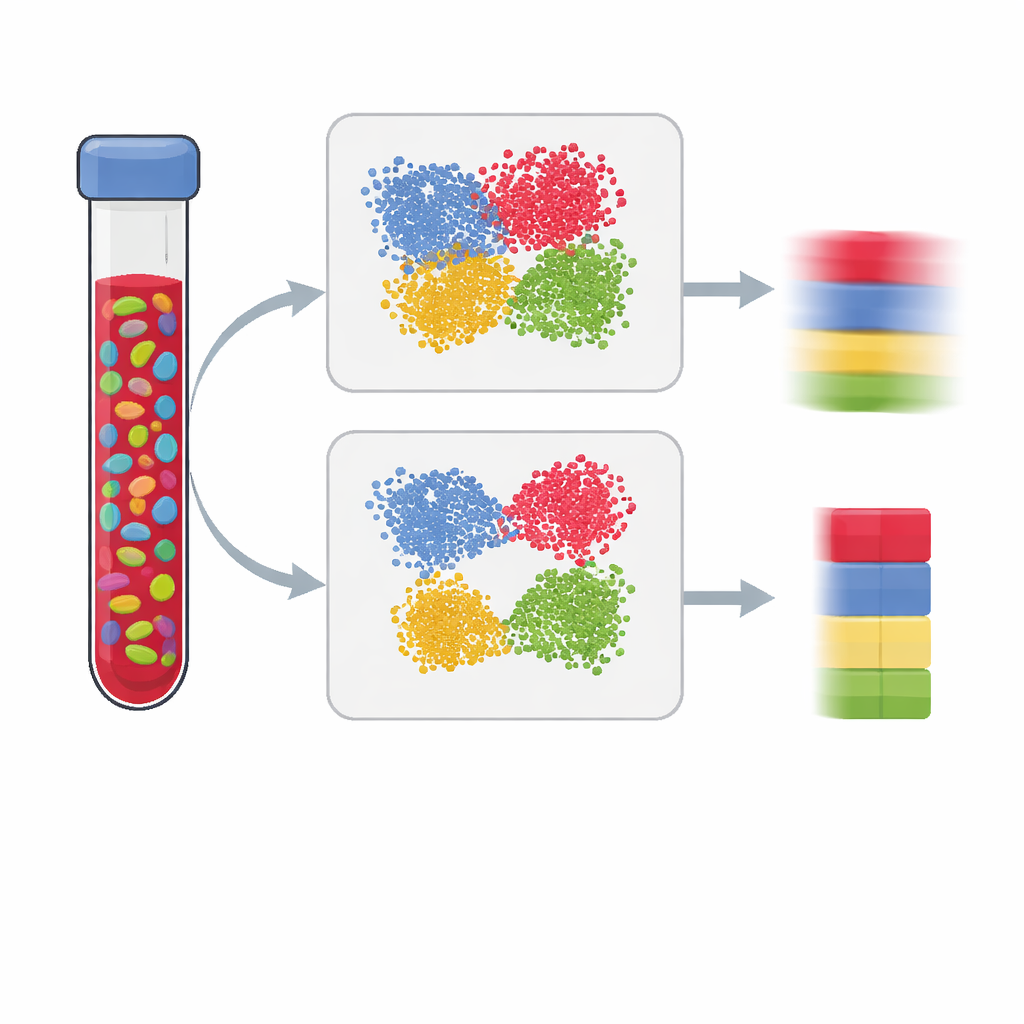
Dlaczego mieszanka komórek ma znaczenie
Badania asocjacji w skali epigenomu poszukują różnic w metylacji DNA — dodawaniu małych grup chemicznych, które regulują aktywność genów — między osobami z daną cechą lub chorobą a tymi bez niej. Ponieważ wzorce metylacji bardzo różnią się między typami komórek, pomiary w materiale zbiorczym, takim jak krew, mogą wprowadzać w błąd: przesunięcie udziału jednego typu komórki odpornościowej na rzecz innego może imitować efekt choroby, nawet jeśli w obrębie poszczególnych typów nic się nie zmieniło. Aby to skorygować, badacze szacują frakcje głównych typów komórek (np. komórki T, B i natural killer) przy użyciu panelu referencyjnego zbudowanego z oczyszczonych komórek lub danych jednokómórkowych. Jakość tego panelu w dużej mierze determinuje, jak dobrze potrafimy „rozmontować” próbkę i jak wiarygodne będą wnioski badania.
Od prostych statystyk do mądrzejszych markerów
Tradycyjnie naukowcy wybierali miejsca w DNA do takich paneli przy użyciu standardowych testów statystycznych. Szukali pozycji, w których dany typ komórki różni się istotnie od pozostałych i sortowali je według statystyki t. W ostatnich latach do ulepszania wyboru stosowano metody optymalizacji i uczenia maszynowego, takie jak IDOL, Elastic Net i Random Forest. Nowe badanie pokazuje, że te podejścia często faworyzują markery o niewielkiej rzeczywistej różnicy między typami komórek, zwłaszcza gdy dostępna jest tylko garść oczyszczonych próbek. Takie markery o „niskim rozmiarze efektu” mogą wyglądać przekonująco na danych treningowych, ale zawodzić w nowych zestawach danych, subtelnie pogarszając dokładność oszacowań udziałów typów komórek.
Znajdowanie wyraźnych przerw między typami komórek
Autorzy proponują bardziej bezpośredni sposób oceny użyteczności markera: „wskaźnik specyficzności przerwy”. Zamiast skupiać się jedynie na istotności statystycznej, wskaźnik ten mierzy, jak czysto miejsce w DNA oddziela dany typ komórki od wszystkich pozostałych, przez analizę przerwy między najwyższą wartością w docelowym typie a najniższą wartością we wszystkich innych komórkach (lub odwrotnie w przypadku niskich wartości). Markery z dużymi dodatnimi przerwami są zarówno specyficzne, jak i odporne. Przy użyciu istniejących danych o komórkach układu odpornościowego badacze wykazali, że ranking markerów wg tego wskaźnika prowadzi do miejsc w DNA o znacznie większych różnicach między typami komórek niż tradycyjna metoda. Panele zbudowane z takich markerów opartych na przerwach dały dokładniejsze oszacowania frakcji komórek w wielu podgrupach odpornościowych, szczególnie w przypadku trudniejszych do rozróżnienia populacji, takich jak pamięciowe komórki CD4 T. 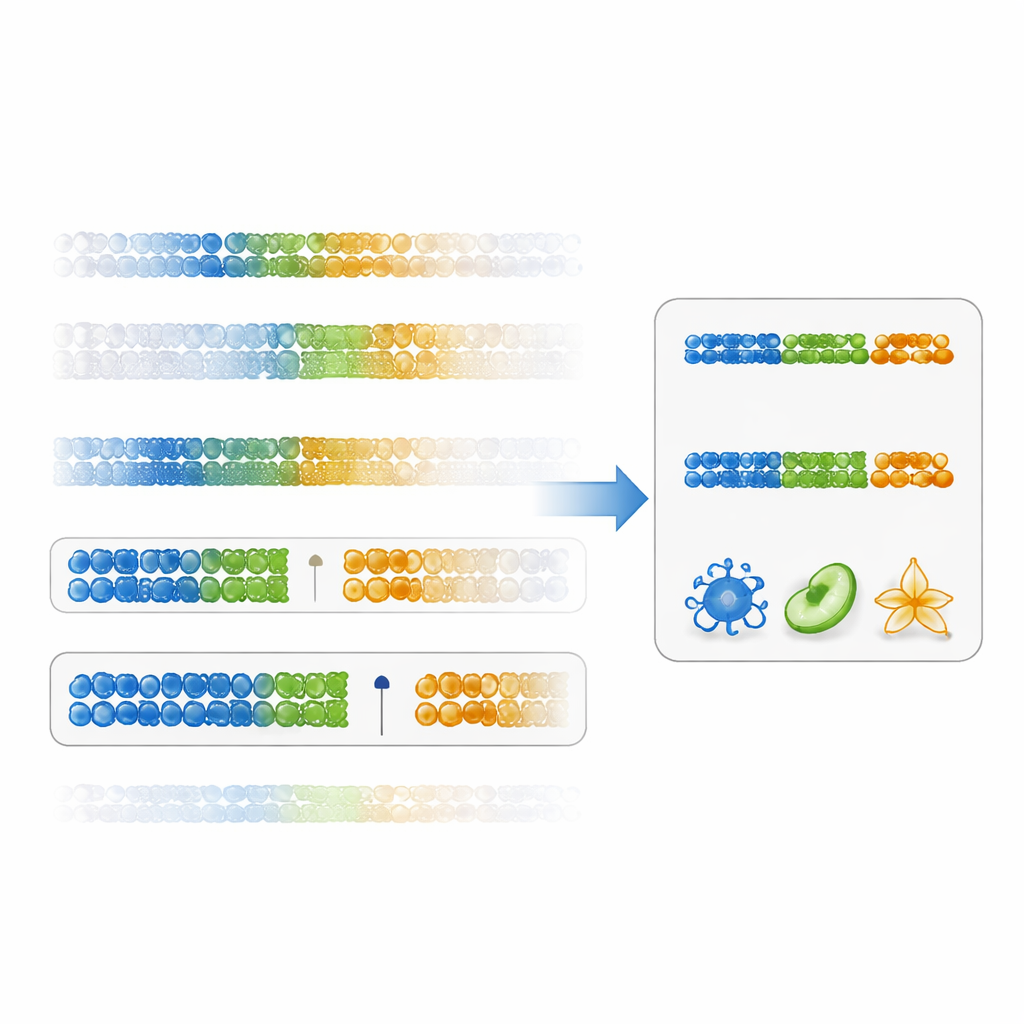
Dlaczego niskie oceny i przeuczenie szkodzą
Zespół sprawdził również, czy zaawansowane narzędzia optymalizacyjne lub modele uczenia maszynowego mogą poprawić ich podejście oparte na przerwach. Zamiast tego stwierdzili przeciwny efekt. Metody takie jak IDOL, Elastic Net i Random Forest miały tendencję do wybierania cech o mniejszych rozmiarach efektu i wypadały gorzej przy ocenie na niezależnych mieszankach lub na prawdziwych próbkach krwi z znanymi liczbami komórek. Sugeruje to, że przy zaledwie kilku tuzinach próbek treningowych złożone modele przeuczają się na nieistotne cechy danych zamiast wychwytywać ogólne wzorce. Natomiast panele zbudowane wyłącznie z silnie hypometylowanych markerów o wysokich wskaźnikach przerwy nie tylko poprawiały dokładność dekonwolucji, ale też lepiej odzwierciedlały znane trendy biologiczne, takie jak wzrost liczby komórek natural killer wraz z wiekiem.
Wyraźniejsze sygnały chorobowe dzięki lepszym panelom
Aby sprawdzić, jak te udoskonalenia wpływają na praktykę, autorzy ponownie przeanalizowali duże badania dotyczące schizofrenii i cukrzycy typu 1. Użycie ich zoptymalizowanych paneli referencyjnych zmieniło oszacowane frakcje komórek tylko nieznacznie, ale te drobne przesunięcia wyostrzyły wyniki kolejnych analiz. Zmiany metylacji związane z chorobą stały się bardziej wzbogacone w ścieżki już powiązane z zapaleniem i autoimmunizacją, a specyficzne geny związane z sygnalizacją odpornościową ujawniły się wyraźniej. Innymi słowy, lepszy dobór markerów redukował szum i sprawiał, że biologiczna narracja była spójniejsza.
Co to oznacza dla przyszłych badań
Dla osób spoza specjalności kluczowy przekaz jest taki, że nie wszystkie statystycznie istotne sygnały są równie użyteczne. Przy rozplątywaniu mieszanych tkanek najważniejsze jest, jak wyraźnie marker odróżnia jeden typ komórki od innego, a nie tylko jak imponująco wygląda jego wartość P. Faworyzując miejsca w DNA z dużymi, czystymi przerwami między typami komórek — zwłaszcza te, które są unikalnie niemetylowane w danym typie — badacze mogą budować bardziej niezawodne panele referencyjne nawet z małych zbiorów danych. Autorzy dodali narzędzia do konstrukcji takich paneli do oprogramowania EpiDISH, co pomoże przyszłym badaniom wyciągać dokładniejsze i biologicznie bardziej sensowne wnioski z danych o metylacji DNA.
Cytowanie: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Słowa kluczowe: metylacja DNA, dekonwolucja typów komórek, epigenomika, komórki odpornościowe, panele referencyjne