Clear Sky Science · pt
Diretrizes para otimizar painéis de referência de metilação do DNA para desconvolução de tipos celulares
Vendo no Interior de Tecidos Mistos
Estudos modernos sobre saúde e doença costumam medir marcas químicas no nosso DNA na tentativa de entender como ambiente e estilo de vida deixam sinais nos genes. Mas a maioria dos testes é feita em tecidos mistos, como o sangue, que contêm muitos tipos celulares. Se não soubermos a proporção de cada tipo celular presente, podemos confundir uma mudança na mistura celular com um verdadeiro sinal de doença. Este artigo explica como construir “painéis de referência” melhores que permitem estimar com precisão as misturas celulares a partir de dados de metilação do DNA, resultando em conclusões mais claras e confiáveis. 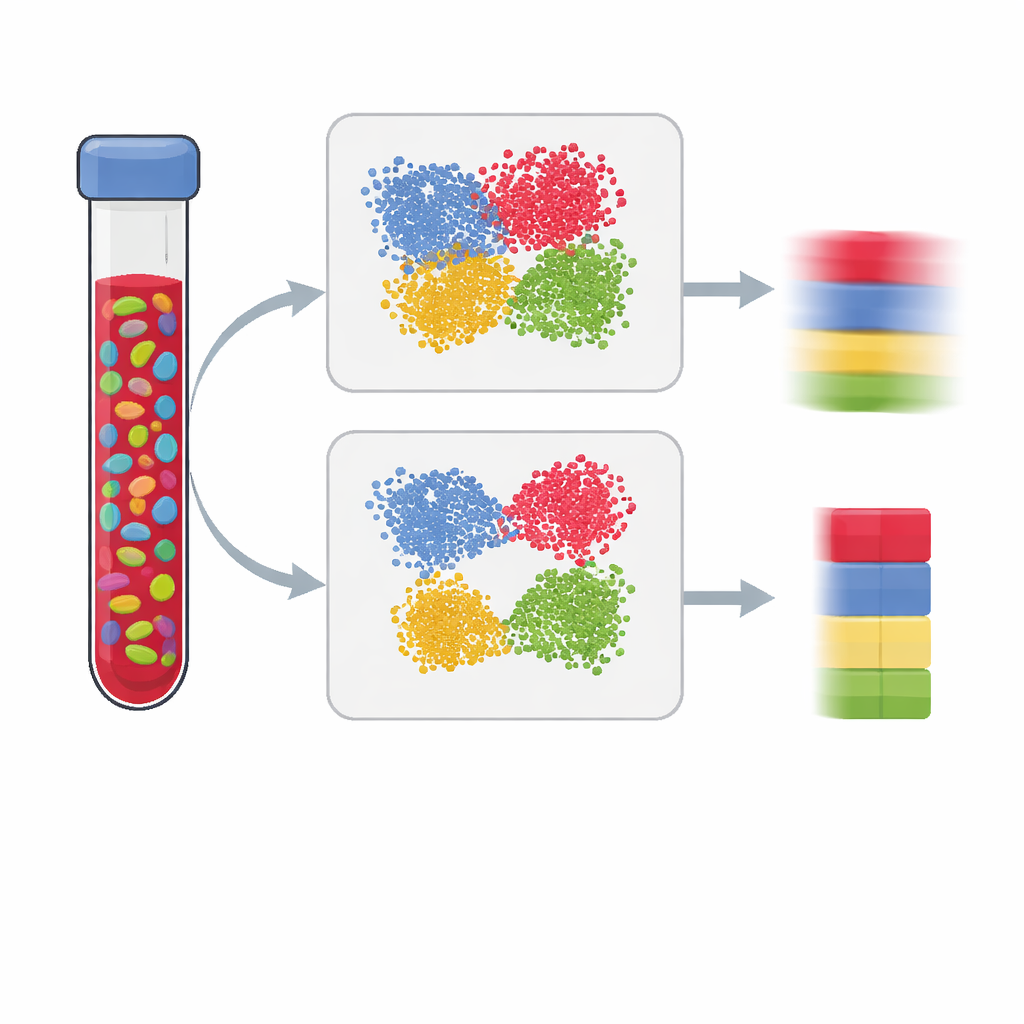
Por que a Mistura Celular Importa
Estudos de associação epigenômica procuram diferenças na metilação do DNA — a adição de pequenas marcas químicas que ajudam a controlar a atividade gênica — entre pessoas com e sem uma característica, como uma doença. Como os padrões de metilação variam muito entre tipos celulares, medições em amostras em massa, como sangue, podem enganar: uma mudança na proporção de um tipo de célula imune em relação a outro pode imitar um efeito de doença mesmo que nada tenha mudado dentro de cada tipo celular. Para corrigir isso, os pesquisadores estimam as frações dos principais tipos celulares (como células T, células B e células NK) usando um painel de referência construído a partir de células purificadas ou de células únicas. A qualidade desse painel determina em grande parte o quão bem podemos “desmisturar” uma amostra e, por consequência, quão confiáveis são as conclusões do estudo.
Da Estatística Simples a Marcadores Mais Inteligentes
Tradicionalmente, cientistas escolhiam sítios de DNA para esses painéis usando testes estatísticos padrão. Procuravam posições onde um tipo celular difere significativamente de todos os outros e as classificavam por um t-estatístico. Mais recentemente, métodos de otimização e aprendizado de máquina como IDOL, Elastic Net e Random Forests têm sido usados para refinar essas escolhas. O novo estudo mostra que essas abordagens frequentemente priorizam marcadores com pequenas diferenças no mundo real entre tipos celulares, especialmente quando há disponível apenas um número reduzido de amostras purificadas. Tais marcadores de “baixo tamanho de efeito” podem parecer convincentes nos dados de treinamento, mas falharem em conjuntos novos, degradando sutilmente a precisão das estimativas de tipos celulares.
Encontrando Lacunas Claras entre Tipos Celulares
Os autores propõem uma maneira mais direta de avaliar a utilidade de um marcador: uma “pontuação de especificidade por lacuna”. Em vez de focar apenas na significância estatística, essa pontuação mede o quão claramente um sítio de DNA separa um tipo celular de todos os outros, observando a lacuna entre o valor mais alto no tipo celular alvo e o valor mais baixo em todos os outros (ou o inverso para valores baixos). Marcadores com grandes lacunas positivas são ao mesmo tempo específicos e robustos. Usando dados existentes de células imunes, os pesquisadores demonstraram que classificar marcadores por essa pontuação produz sítios de DNA com diferenças entre tipos celulares muito maiores do que o método tradicional. Painéis construídos a partir desses marcadores baseados em lacuna geraram estimativas de fração celular mais precisas em muitos subconjuntos imunes, particularmente para populações mais difíceis de resolver, como células T CD4 de memória. 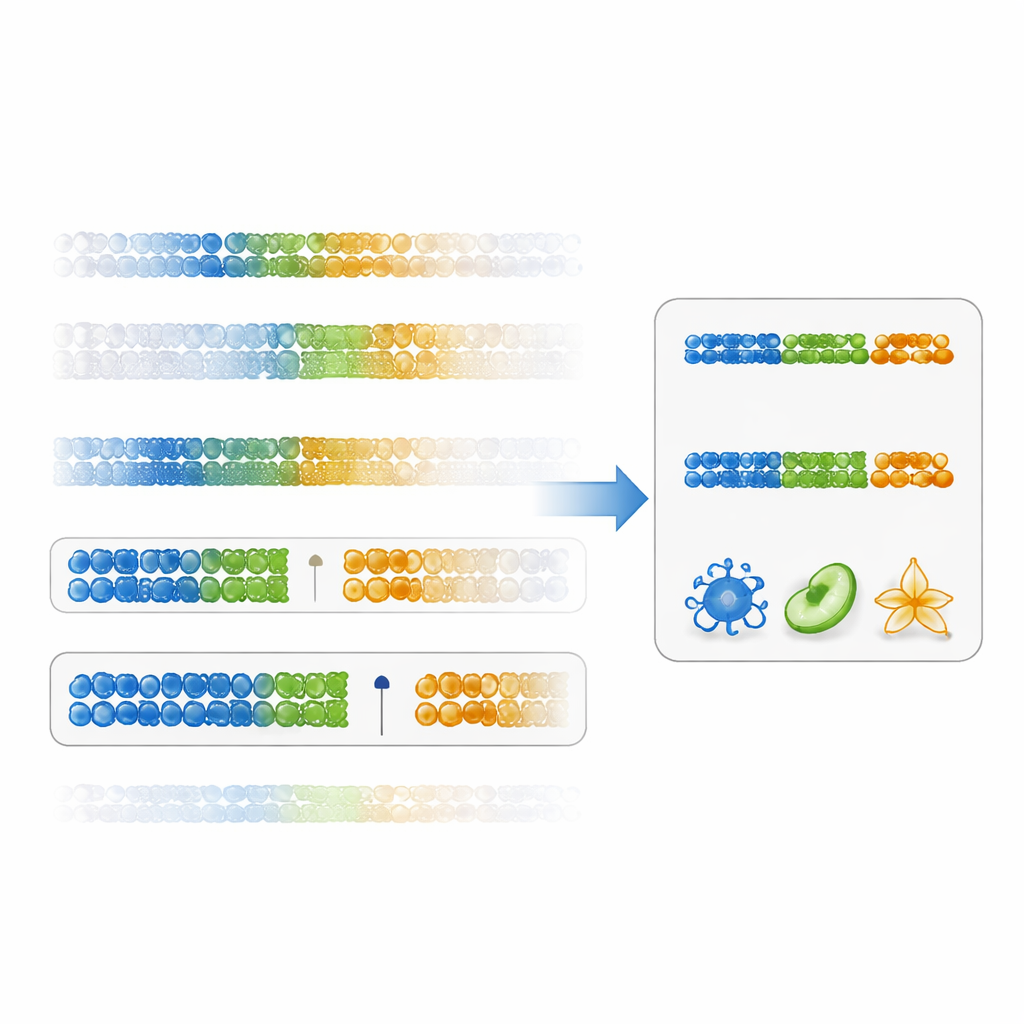
Por que Marcadores Fracos e Overfitting Prejudicam
A equipe também testou se ferramentas de otimização avançadas ou modelos de aprendizado de máquina poderiam melhorar a abordagem baseada em lacunas. Em vez disso, encontraram o oposto. Métodos como IDOL, Elastic Net e Random Forest tenderam a selecionar características com menores tamanhos de efeito e tiveram desempenho pior quando avaliados em misturas independentes ou em amostras de sangue reais com contagens celulares conhecidas. Isso sugere que, com apenas algumas dezenas de amostras de treinamento, modelos complexos se ajustam demais a peculiaridades dos dados em vez de capturar padrões gerais. Em contraste, painéis construídos exclusivamente a partir de marcadores fortemente hipometilados com altas pontuações de lacuna não só melhoraram a precisão da desconvolução, mas também refletiram melhor tendências biológicas conhecidas, como o aumento com a idade das células NK.
Sinais de Doença Mais Claros com Melhores Painéis
Para ver como essas melhorias se manifestam na prática, os autores reanalisaram grandes estudos de esquizofrenia e diabetes tipo 1. Usar seus painéis de referência otimizados alterou apenas ligeiramente as frações celulares estimadas, mas essas pequenas mudanças afinaram as descobertas subsequentes. Alterações associadas à doença na metilação tornaram-se mais enriquecidas em vias já implicadas em inflamação e autoimunidade, e genes específicos ligados ao sinal imunológico emergiram com mais clareza. Em outras palavras, uma seleção de marcadores melhor reduziu o ruído e tornou a história biológica mais coerente.
O Que Isso Significa para Estudos Futuros
Para não especialistas, a mensagem principal é que nem todos os sinais estatisticamente significativos são igualmente úteis. Ao tentar desvendar tecidos mistos, o que mais importa é o quão claramente um marcador distingue um tipo celular de outro, não apenas quão impressionante parece seu valor de P. Favorecendo sítios de DNA com lacunas grandes e limpas entre tipos celulares — especialmente aqueles que são unicamente pouco metilados em um dado tipo — os pesquisadores podem construir painéis de referência mais confiáveis mesmo a partir de conjuntos de dados pequenos. Os autores adicionaram ferramentas para construir esses painéis ao software EpiDISH, ajudando futuros estudos a extrair conclusões mais precisas e biologicamente significativas a partir de dados de metilação do DNA.
Citação: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Palavras-chave: metilação do DNA, desconvolução de tipos celulares, epigenômica, células imunes, painéis de referência