Clear Sky Science · de
Richtlinien zur Optimierung von DNA‑Methylierungs‑Referenzpanels für die Zelltyp‑Deconvolution
Ein Blick in gemischte Gewebe
Moderne Studien zu Gesundheit und Krankheit messen häufig chemische Markierungen auf unserer DNA, um zu verstehen, wie Umwelt und Lebensstil Spuren an unserem Erbgut hinterlassen. Viele Messungen erfolgen jedoch an gemischten Geweben wie Blut, das zahlreiche Zelltypen enthält. Können wir nicht bestimmen, welcher Anteil welcher Zelltyp hat, besteht die Gefahr, dass eine Veränderung in der Zellzusammensetzung fälschlich als biologisches Krankheitszeichen interpretiert wird. Dieser Artikel erklärt, wie man bessere „Referenzpanels“ erstellt, mit denen Wissenschaftler die Zellmischungen aus DNA‑Methylierungsdaten präzise schätzen können, was zu klareren und verlässlicheren Ergebnissen führt. 
Warum die Zellmischung wichtig ist
Epigenomweite Assoziationsstudien suchen nach Unterschieden in der DNA‑Methylierung – der Anlagerung kleiner chemischer Gruppen, die die Genaktivität mitsteuern – zwischen Personen mit und ohne ein Merkmal, etwa einer Krankheit. Da Methylierungsmuster zwischen Zelltypen stark variieren, kann eine Messung im Bulk‑Blut irreführend sein: Ein Wechsel von einem Immunzelltyp zu einem anderen kann einen krankheitsbedingten Effekt nachahmen, selbst wenn sich innerhalb der einzelnen Zelltypen nichts geändert hat. Um dies zu korrigieren, schätzen Forscher die Anteile der wichtigsten Zelltypen (z. B. T‑Zellen, B‑Zellen und natürliche Killerzellen) mithilfe eines Referenzpanels, das aus gereinigten Zellen oder Einzelzellmessungen aufgebaut ist. Die Qualität dieses Panels bestimmt weitgehend, wie gut sich eine Probe „entmischen“ lässt und somit wie verlässlich die Studienergebnisse sind.
Von einfachen Statistiken zu schlaueren Markern
Traditionell wählten Wissenschaftler DNA‑Positionen für solche Panels mithilfe standardmäßiger statistischer Tests aus. Sie suchten nach Stellen, an denen sich ein Zelltyp signifikant von allen anderen unterschied, und ordneten diese nach einem t‑Statistik‑Wert. Neuerdings wurden Optimierungs‑ und Machine‑Learning‑Methoden wie IDOL, Elastic Net und Random Forest eingesetzt, um diese Auswahl zu verfeinern. Die neue Studie zeigt, dass diese Ansätze häufig Marker priorisieren, deren reale Unterschiede zwischen Zelltypen klein sind, insbesondere wenn nur wenige gereinigte Proben zur Verfügung stehen. Solche Marker mit geringer Effektstärke wirken in den Trainingsdaten überzeugend, können aber in neuen Datensätzen versagen und dadurch die Genauigkeit der Zelltyp‑Schätzungen unterschwellig verschlechtern.
Klar erkennbare Abstände zwischen Zelltypen finden
Die Autorinnen und Autoren schlagen eine direktere Methode vor, um die Nützlichkeit eines Markers zu beurteilen: einen „Gap‑Spezifitäts‑Score“. Anstatt sich nur auf statistische Signifikanz zu konzentrieren, misst dieser Score, wie sauber eine DNA‑Stelle einen Zelltyp von allen anderen trennt, indem er die Lücke zwischen dem höchsten Wert im Zielzelltyp und dem niedrigsten Wert in allen anderen Zellen betrachtet (oder umgekehrt bei niedrigen Werten). Marker mit großen positiven Lücken sind sowohl spezifisch als auch robust. Anhand vorhandener Immunzell‑Daten zeigten die Forschenden, dass eine Rangfolge nach diesem Score DNA‑Stellen mit wesentlich größeren Unterschieden zwischen Zelltypen liefert als die herkömmliche Methode. Panels, die aus diesen gap‑basierten Markern gebaut wurden, lieferten über viele Immununtergruppen hinweg genauere Zellanteilsschätzungen, insbesondere für schwer zu trennende Populationen wie Gedächtnis‑CD4‑T‑Zellen. 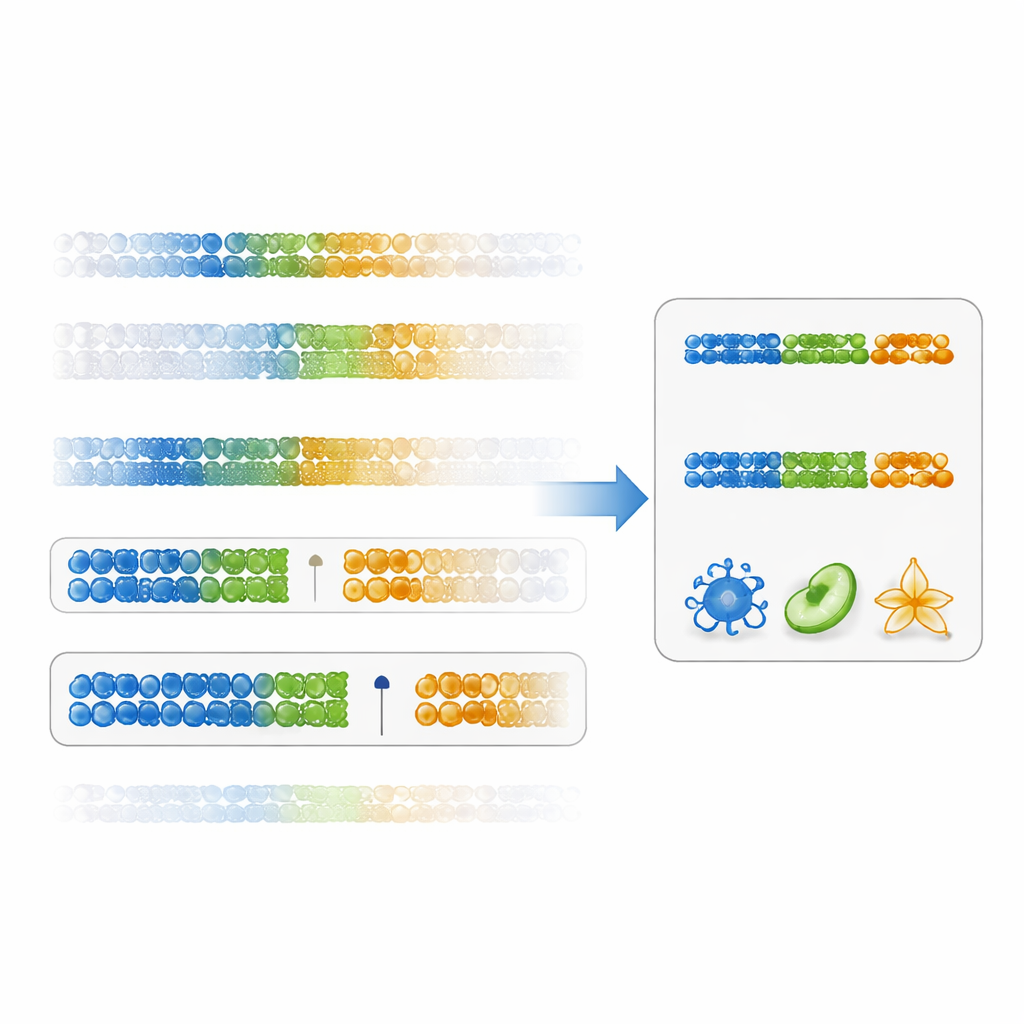
Warum geringe Markersignale und Overfitting schaden
Das Team prüfte außerdem, ob fortgeschrittene Optimierungswerkzeuge oder Machine‑Learning‑Modelle den gap‑basierten Ansatz verbessern könnten. Stattdessen fanden sie das Gegenteil. Methoden wie IDOL, Elastic Net und Random Forest neigten dazu, Merkmale mit kleineren Effektstärken auszuwählen und schnitten bei der Bewertung an unabhängigen Mischproben oder an echten Blutproben mit bekannten Zellzahlen schlechter ab. Das legt nahe, dass komplexe Modelle bei nur einigen Dutzend Trainingsproben Besonderheiten der Daten überanpassen, statt allgemeine Muster zu erfassen. Im Gegensatz dazu verbesserten Panels, die ausschließlich aus stark hypomethylierten Markern mit hohen Gap‑Scores bestanden, nicht nur die Deconvolution‑Genauigkeit, sondern erfassten auch besser bekannte biologische Trends, wie den altersbedingten Anstieg der natürlichen Killerzellen.
Klarere Krankheitsbefunde durch bessere Panels
Um zu zeigen, wie sich diese Verbesserungen in der Praxis auswirken, reanalysierten die Autorinnen und Autoren große Studien zu Schizophrenie und Typ‑1‑Diabetes. Mit ihren optimierten Referenzpanels änderten sich die geschätzten Zellanteile nur geringfügig, doch diese kleinen Verschiebungen schärften die nachfolgenden Befunde. Krankheitsassoziierte Methylierungsänderungen wurden stärker in Signalwegen angereichert, die bereits mit Entzündung und Autoimmunität in Verbindung stehen, und spezifische Gene der Immun‑Signalgebung traten klarer hervor. Anders gesagt: Bessere Markerwahl reduzierte Rauschen und machte die biologische Geschichte kohärenter.
Was das für künftige Studien bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft: Nicht alle statistisch signifikanten Signale sind gleichermaßen nützlich. Beim Entwirren gemischter Gewebe kommt es vor allem darauf an, wie klar ein Marker einen Zelltyp von anderen unterscheidet, nicht nur darauf, wie beeindruckend sein P‑Wert wirkt. Indem man DNA‑Stellen mit großen, sauberen Lücken zwischen Zelltypen bevorzugt—insbesondere solche, die in einem bestimmten Zelltyp eindeutig unmethyliert sind—können Forschende auch aus kleinen Datensätzen verlässlichere Referenzpanels bauen. Die Autorinnen und Autoren haben Werkzeuge zur Konstruktion solcher Panels in die Software EpiDISH integriert, um künftigen Studien zu helfen, aus DNA‑Methylierungsdaten genauere und biologisch aussagekräftigere Schlüsse zu ziehen.
Zitation: Guo, X., Teschendorff, A.E. Guidelines on optimizing DNA methylation reference panels for cell-type deconvolution. Commun Biol 9, 454 (2026). https://doi.org/10.1038/s42003-026-09745-1
Schlüsselwörter: DNA‑Methylierung, Zelltyp‑Deconvolution, Epigenomik, Immunzellen, Referenzpanels