Clear Sky Science · sv
Q-CaDD: accelerera in silico-metoder med kvantberäkning och maskininlärning för Epidermal tillväxtfaktorreceptorn
Varför nya datorverktyg är viktiga för framtida cancerläkemedel
Att utforma nya läkemedel är lite som att leta efter en nål i en höstack av möjliga molekyler. För cancerformer som drivs av ett protein kallat Epidermal Growth Factor Receptor (EGFR) måste forskare hitta föreningar som binder detta protein hårt men samtidigt är säkra för patienter. Denna artikel presenterar Q-CaDD, ett datorbaserat ramverk som blandar dagens maskininlärning med framväxande idéer inom kvantberäkning för att sållra hundratusentals kandidatmolekyler mer effektivt och för att markera dem som kan bli säkrare, mer verksamma läkemedel.
Från ett cancerkopplat protein till ett digitalt sökproblem
EGFR sitter på cellernas yta och hjälper till att styra hur de växer och delar sig. När det fungerar felaktigt, vilket ofta sker vid icke-småcellig lungcancer, kan celler dela sig okontrollerat. Läkemedel som blockerar EGFR finns redan, men tumörer kan utveckla resistens och inte alla patienter svarar bra. Istället för att testa nya föreningar en och en i labbet använder Q-CaDD computersimuleringar för att utforska kemiskt utrymme i stor skala, och söker efter molekyler som både fäster vid EGFR och uppvisar tecken på låg toxicitet. Detta tillvägagångssätt syftar till att göra de tidiga stegen i läkemedelsupptäckt snabbare, billigare och mer riktade.
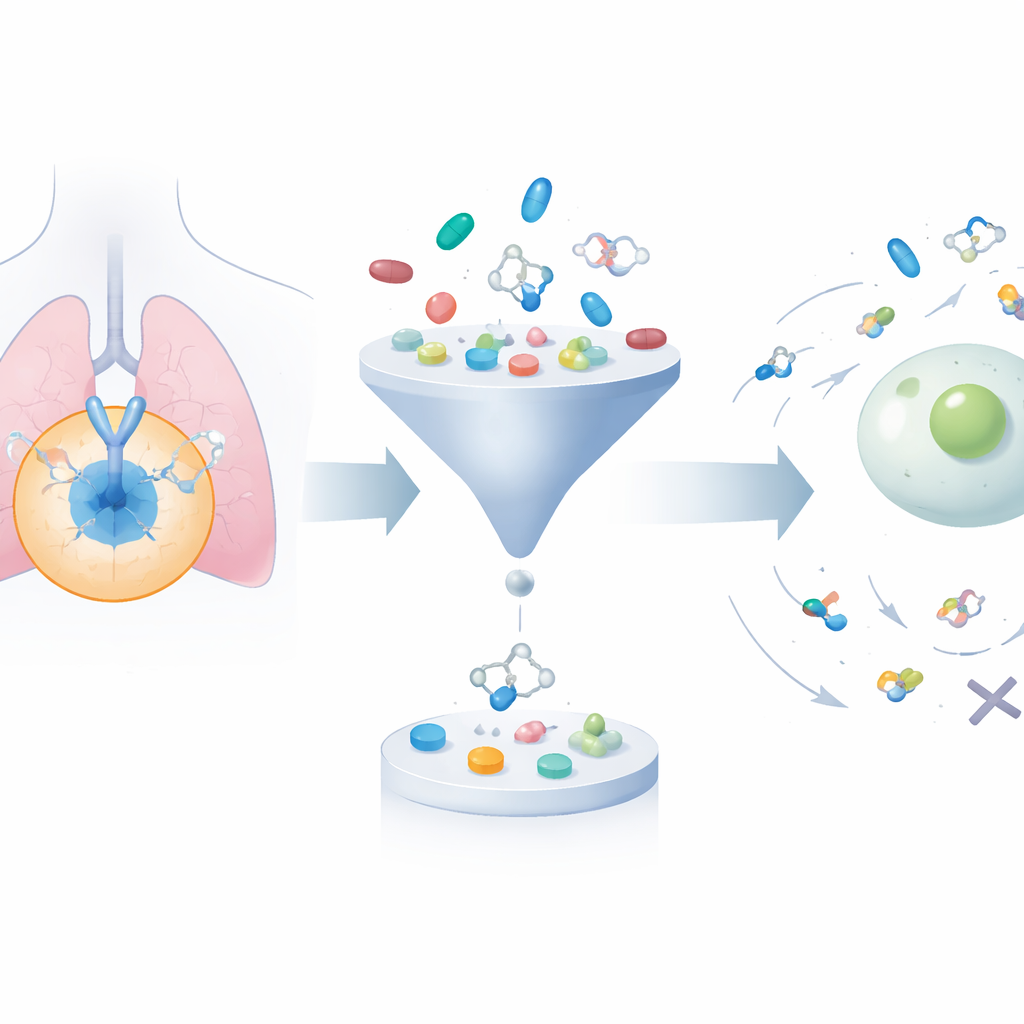
Att växa och trimma ett omfångsrikt bibliotek av molekyler
Ramverket börjar med att samla in omkring 24 000 kända EGFR-blockerande molekyler från offentliga databaser. Därefter använder det en generativ algoritm för att systematiskt modifiera deras strukturer och producera ungefär 200 000 relaterade kandidater. Två väl etablerade filter för ”läkemedelslikhet” tillämpas för att sålla bort föreningar som är för klumpiga, för fettlösliga eller på annat sätt osannolika att bete sig väl i kroppen, vilket minskar mängden till färre än 50 000. Därefter använder ett dockningsprogram en virtuell passform för varje molekyl i den tredimensionella fickan på EGFR där verkliga läkemedel skulle binda, och uppskattar hur starkt varje förening kan fästa. Detta fokuserar uppmärksamheten på föreningar som både är kemiskt rimliga och förutsagda att interagera väl med målet.
Lära datorer att känna igen toxikologiska varningssignaler
Bindning till EGFR är bara halva berättelsen; en lovande förening måste också undvika att skada frisk vävnad. För att uppskatta toxicitet använder studien en stor offentlig dataset kallad Tox21, som registrerar hur över 10 000 kemikalier påverkar olika cellulära signalvägar. Författarna fokuserar på en väg kopplad till androgenreceptorn, vald eftersom den är väl annoterad och biologiskt relevant för flera cancerformer. Varje Tox21-molekyl översätts till ett numeriskt fingeravtryck som fångar dess strukturella egenskaper och likheter med andra kemikalier. Dessa fingeravtryck matar flera prediktiva modeller, inklusive neurala nätverk, beslutsträd, en traditionell supportvektormaskin och en kvantinspirerad supportvektormaskin som använder en enkel kvantkrets för att jämföra föreningar i ett annat matematiskt rum.
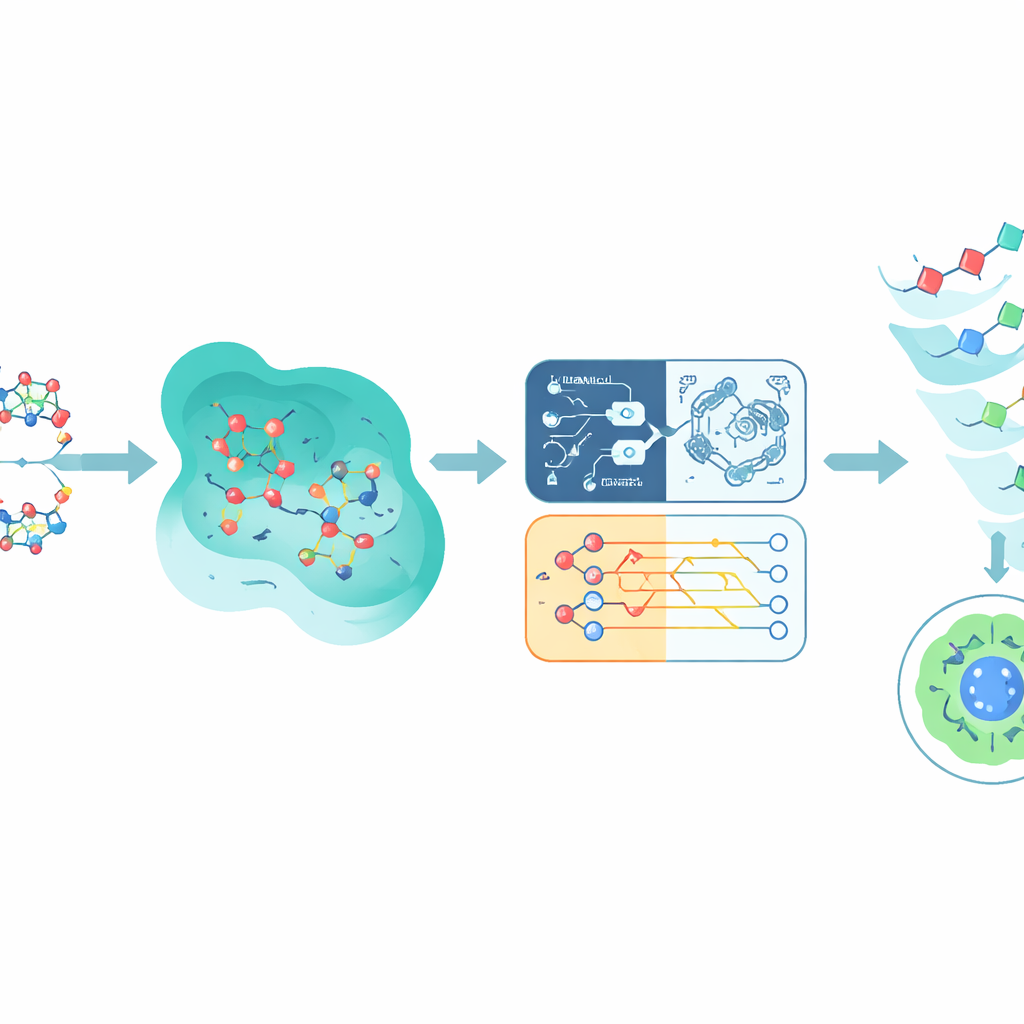
Att blanda kvant- och klassiska prediktioner
I stället för att satsa på en enda modell kombinerar Q-CaDD utsignalerna från alla fyra i ett ensemble, där störst vikt ges till det neurala nätverket men där även den svagare men distinkta signalen från den kvantinspirerade modellen tas med. När metoden testas på tidigare osedd Tox21-data presterar detta blandade tillvägagångssätt bättre än någon enskild modell för att skilja mer och mindre toxiska föreningar, mätt med en standardiserad rankningspoäng kallad area under ROC-kurvan. Även om förbättringen är måttlig och den kvantdel fortfarande körs i en simulator snarare än på en verklig kvantchip, tyder resultaten på att kvantinspirerade metoder kan tillföra användbar nyans till befintliga maskininlärningspipelines även i ett tidigt skede.
Från datorpoäng till framtida labbtester
Efter att ha validerat toxicitetsmodellerna applicerar författarna Q-CaDD:s fullständiga pipeline på det filtrerade EGFR-fokuserade biblioteket. De undviker att göra ett hårt ja-eller-nej-beslut om toxicitet, i stället behåller de kontinuerliga riskpoäng och kombinerar dem med dockningsuppskattningar av bindningsstyrka. Detta ger en prioriteringslista över kandidatmolekyler, varav några ser ut att binda EGFR starkare än ett referensläkemedel samtidigt som de behåller låg förutsagd toxicitet. Dessa molekyler påstås inte vara nya läkemedel; de markeras som ledtrådar som förtjänar laboratorietester. Studiens huvudslutsats för icke-specialister är inte att kvantdatorer redan har revolutionerat läkemedelsupptäckt, utan att noggrant utformade hybrider av klassiska och kvantinspirerade verktyg redan kan hjälpa till att skärpa sökningen, peka forskare mot bättre läkemedelskandidater snabbare samtidigt som man är realistisk om dagens hårdvarubegränsningar.
Citering: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Nyckelord: kvantbaserad läkemedelsupptäckt, EGFR-hämmare, maskininlärning toxicitet, virtuell screening, icke-småcellig lungcancer