Clear Sky Science · it
Q-CaDD: accelerare le metodologie in silico con calcolo quantistico e apprendimento automatico per il recettore del fattore di crescita epidermico
Perché nuovi strumenti informatici contano per i farmaci antitumorali futuri
Progettare nuovi medicinali è un po’ come cercare un ago in un pagliaio di possibili molecole. Per i tumori guidati da una proteina chiamata Recettore del Fattore di Crescita Epidermico (EGFR), i ricercatori devono trovare composti che si leghino a questa proteina con forza ma che restino sicuri per i pazienti. Questo articolo presenta Q-CaDD, un quadro computazionale che fonde l’apprendimento automatico odierno con idee emergenti del calcolo quantistico per setacciare centinaia di migliaia di molecole candidate in modo più efficiente e segnalare quelle che potrebbero diventare farmaci più sicuri ed efficaci.
Da una proteina legata al cancro a un problema di ricerca digitale
EGFR si trova sulla superficie delle cellule e aiuta a controllare come crescono e si dividono. Quando funziona male, come spesso accade nel carcinoma polmonare non a piccole cellule, le cellule possono moltiplicarsi incontrollabilmente. Esistono già farmaci che bloccano EGFR, ma i tumori possono diventare resistenti e non tutti i pazienti rispondono bene. Invece di testare nuovi composti uno per uno in laboratorio, Q-CaDD usa simulazioni al computer per esplorare in massa lo spazio chimico, cercando molecole che si leghino a EGFR e mostrino segnali di bassa tossicità. Questo approccio punta a rendere i primi passi della scoperta di farmaci più rapidi, economici e guidati.
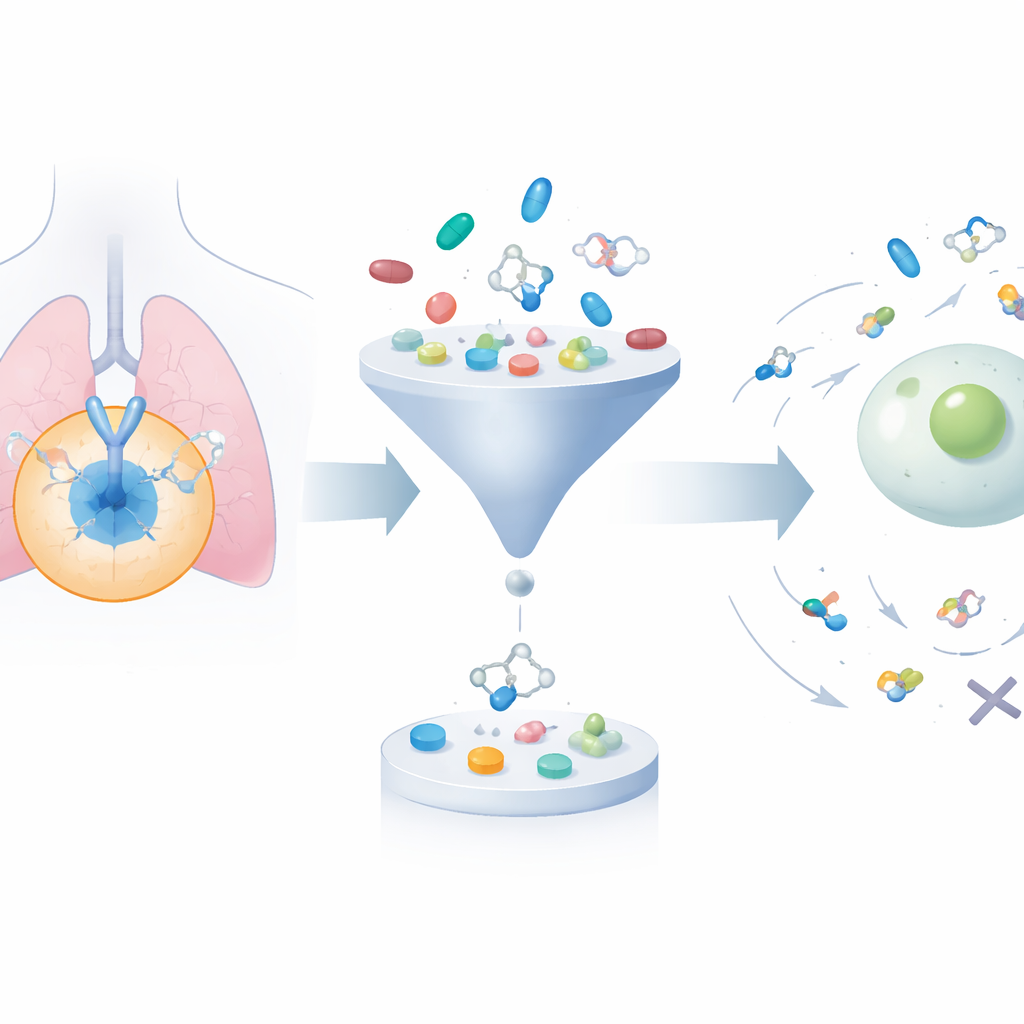
Espandere e potare una vasta libreria di molecole
Il quadro inizia raccogliendo circa 24.000 molecole note per bloccare EGFR da banche dati pubbliche. Poi utilizza un algoritmo generativo per modificare sistematicamente le loro strutture, producendo all’incirca 200.000 candidati correlati. Vengono applicati due filtri consolidati di “drug-likeness” per eliminare composti troppo voluminosi, troppo lipofili o altrimenti improbabili a comportarsi bene nell’organismo, riducendo l’insieme a meno di 50.000. Successivamente, un programma di docking adatta virtualmente ogni molecola nella tasca tridimensionale di EGFR dove i farmaci reali si legherebbero, stimando quanto forte potrebbe essere ogni legame. Questo restringe l’attenzione a composti che sono sia chimicamente ragionevoli sia previsti interagire bene con il target.
Addestrare i computer a riconoscere segnali di tossicità
Legarsi a EGFR è solo metà della storia; un composto promettente deve anche evitare di danneggiare i tessuti sani. Per stimare la tossicità, lo studio si rivolge a un ampio dataset pubblico chiamato Tox21, che registra come oltre 10.000 chimici influenzano varie vie cellulari. Gli autori si concentrano su una via collegata al recettore degli androgeni, scelta perché è ben annotata e biologicamente rilevante per diversi tumori. Ogni molecola di Tox21 viene tradotta in un’impronta numerica che cattura le sue caratteristiche strutturali e le somiglianze con altre sostanze chimiche. Queste impronte alimentano diversi modelli predittivi, inclusi reti neurali, alberi decisionali, una tradizionale macchina a vettori di supporto e una macchina a vettori di supporto ispirata al quantistico che usa un semplice circuito quantistico per confrontare i composti in uno spazio matematico differente.
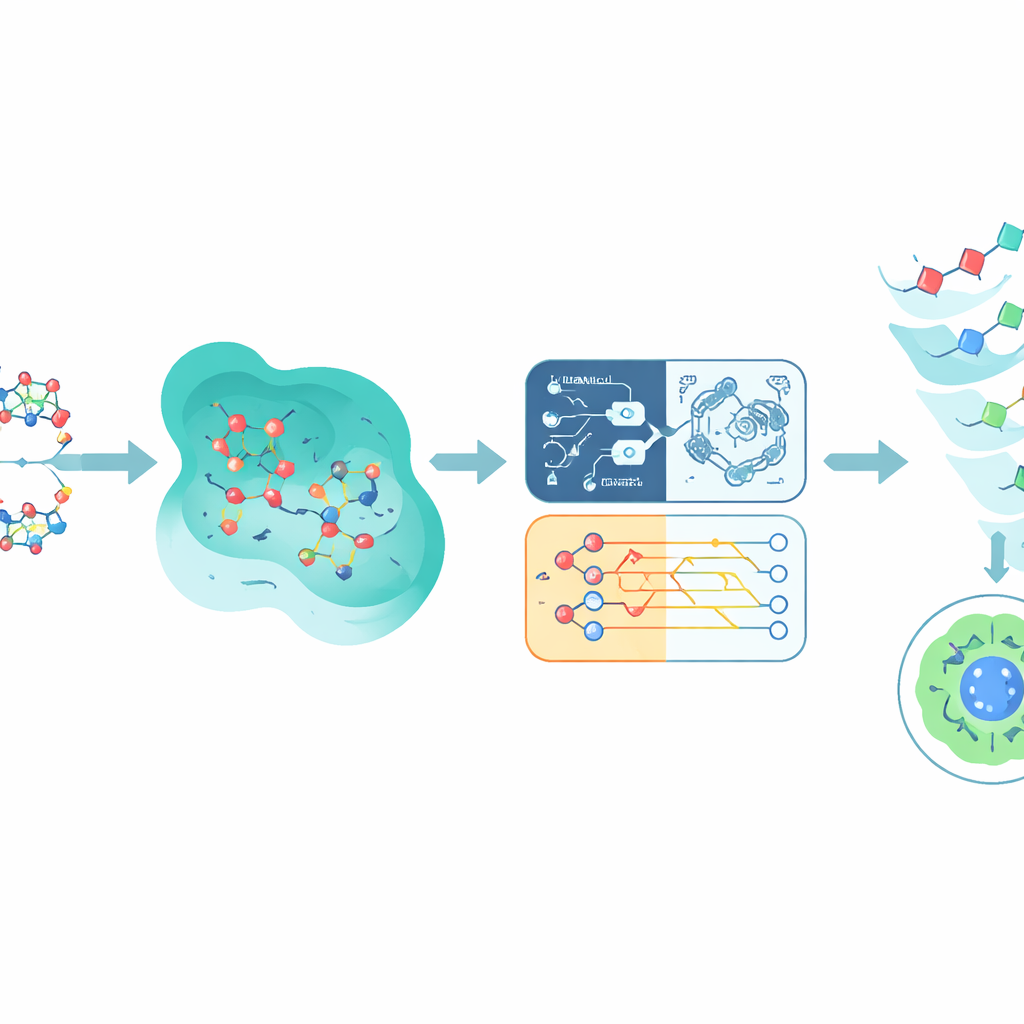
Fondere predizioni quantistiche e classiche
Piuttosto che puntare su un singolo modello, Q-CaDD combina le uscite di tutti e quattro in un ensemble, dando il peso maggiore alla rete neurale ma incorporando comunque il segnale più debole ma distinto del modello quantistico. Testato su dati Tox21 non visti in precedenza, questo approccio ibrido supera ciascun modello individuale nel distinguere composti più o meno tossici, misurato tramite una metrica standard nota come area sotto la curva ROC. Sebbene il miglioramento sia modesto e la parte quantistica venga ancora eseguita su un simulatore piuttosto che su un chip quantistico reale, i risultati suggeriscono che metodi ispirati al quantistico possono aggiungere una sfumatura utile alle pipeline di apprendimento automatico esistenti già nelle fasi iniziali.
Dai punteggi computazionali ai futuri test di laboratorio
Dopo aver convalidato i modelli di tossicità, gli autori applicano l’intera pipeline di Q-CaDD alla libreria filtrata focalizzata su EGFR. Evitano di emettere un giudizio netto sì/no sulla tossicità, preferendo mantenere punteggi di rischio continui e combinarli con le stime di binding ottenute dal docking. Questo produce una lista di priorità di molecole candidate, alcune delle quali sembrano legarsi a EGFR più fortemente di un farmaco di riferimento mantenendo bassa la tossicità prevista. Queste molecole non sono presentate come nuovi medicinali; sono segnalate come lead che meritano test in laboratorio. Il messaggio principale dello studio, per i non specialisti, non è che i computer quantistici abbiano già rivoluzionato la scoperta di farmaci, ma che ibridi ben progettati di strumenti classici e ispirati al quantistico possono già aiutare a rendere la ricerca più mirata, indicando ai ricercatori candidati migliori più rapidamente pur restando realistici sui limiti dell’hardware attuale.
Citazione: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Parole chiave: scoperta di farmaci quantistica, inibitori EGFR, tossicità e apprendimento automatico, screening virtuale, carcinoma polmonare non a piccole cellule