Clear Sky Science · pl
Q-CaDD: przyspieszanie metod in silico za pomocą obliczeń kwantowych i uczenia maszynowego dla receptora naskórkowego czynnika wzrostu
Dlaczego nowe narzędzia komputerowe mają znaczenie dla przyszłych leków przeciwnowotworowych
Projektowanie nowych leków przypomina szukanie igły w stogu siana możliwych cząsteczek. W przypadku nowotworów związanych z białkiem zwanym receptorem naskórkowego czynnika wzrostu (EGFR) badacze muszą znaleźć związki, które mocno wiążą to białko, a jednocześnie są bezpieczne dla pacjentów. W artykule przedstawiono Q-CaDD, komputerowe ramy łączące współczesne uczenie maszynowe z wschodzącymi pomysłami obliczeń kwantowych, służące do wydajniejszego przeszukiwania setek tysięcy kandydatów i wskazywania tych, które mogą stać się bezpieczniejszymi, skuteczniejszymi lekami.
Od białka związanego z rakiem do problemu przeszukiwania w formie cyfrowej
EGFR znajduje się na powierzchni komórek i pomaga kontrolować ich wzrost oraz podziały. Gdy działa nieprawidłowo, jak często ma to miejsce w niedrobnokomórkowym raku płuca, komórki mogą mnożyć się bez kontroli. Leki blokujące EGFR już istnieją, ale nowotwory mogą się uodpornić, a nie każdy pacjent reaguje dobrze. Zamiast testować nowe związki pojedynczo w laboratorium, Q-CaDD wykorzystuje symulacje komputerowe do masowego eksplorowania przestrzeni chemicznej, poszukując cząsteczek, które zarówno przyczepiają się do EGFR, jak i wykazują oznaki niskiej toksyczności. Podejście to ma na celu uczynienie wczesnych etapów odkrywania leków szybszymi, tańszymi i bardziej ukierunkowanymi.
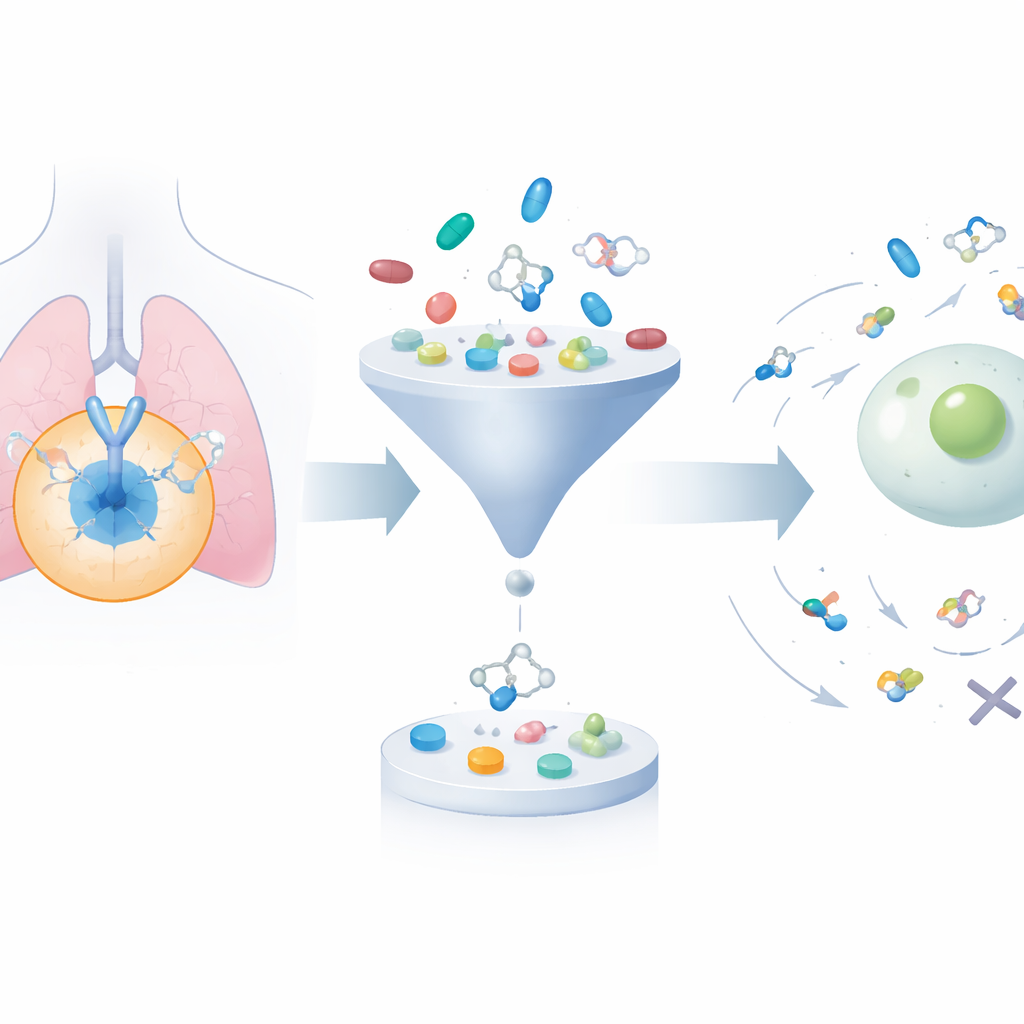
Rozszerzanie i przycinanie ogromnej biblioteki cząsteczek
Ramy rozpoczynają się od zebrania około 24 000 znanych cząsteczek blokujących EGFR z publicznych baz danych. Następnie używany jest algorytm generatywny do systematycznego modyfikowania ich struktur, co daje około 200 000 spokrewnionych kandydatów. Stosuje się dwa dobrze znane filtry „podobieństwa do leku”, aby odsiać związki zbyt masywne, zbyt lipofilne lub w inny sposób mało obiecujące w kontekście zachowania w organizmie, redukując zbiór do poniżej 50 000. Kolejno program dokujący wirtualnie dopasowuje każdą cząsteczkę do trójwymiarowej kieszeni EGFR, w której realnie wiążą się leki, szacując, jak silnie dana cząsteczka może przylegać. To zawęża uwagę do związków zarówno chemicznie rozsądnych, jak i przewidywanych jako dobrze oddziałujące z celem.
Nauczanie komputerów rozpoznawania toksycznych sygnałów ostrzegawczych
Wiązanie z EGFR to tylko połowa historii; obiecujący związek musi też unikać szkody dla zdrowych tkanek. Aby oszacować toksyczność, badanie korzysta z dużego publicznego zestawu danych nazwanego Tox21, który dokumentuje, jak ponad 10 000 chemikaliów wpływa na różne szlaki komórkowe. Autorzy skupili się na jednym szlaku związanym z receptorem androgenowym, wybranym ze względu na dobrą adnotację i biologiczne powiązanie z kilkoma rodzajami nowotworów. Każda cząsteczka z Tox21 jest tłumaczona na numeryczny odcisk palca (fingerprint), który uchwyca jej cechy strukturalne i podobieństwa do innych związków. Te odciski zasilają kilka modeli predykcyjnych: sieci neuronowe, drzewa decyzyjne, tradycyjny wektor maszyn wspierających (SVM) oraz inspirowany kwantowo SVM, wykorzystujący prosty obwód kwantowy do porównywania związków w innym przestrzennie-matematycznym wymiarze.
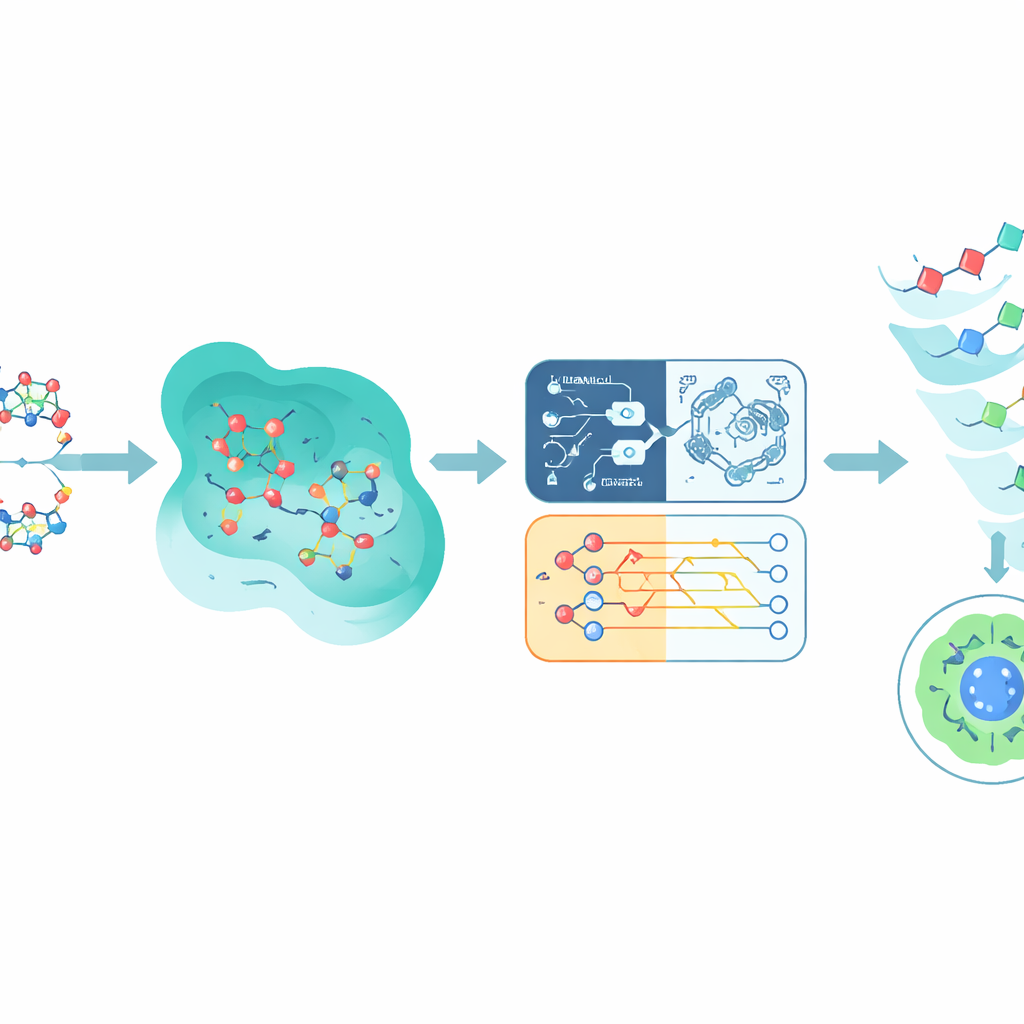
Mieszanie predykcji kwantowych i klasycznych
Zamiast postawić na pojedynczy model, Q-CaDD łączy wyniki wszystkich czterech w zespole modeli (ensemble), nadając największą wagę sieci neuronowej, jednocześnie uwzględniając słabszy, lecz odrębny sygnał z modelu inspirowanego kwantowo. W testach na uprzednio nieznanych danych Tox21 to połączenie przewyższało każdy pojedynczy model w rozróżnianiu związków bardziej i mniej toksycznych, mierzonym standardowym wskaźnikiem AUC (obszar pod krzywą ROC). Chociaż poprawa jest skromna, a część kwantowa wciąż działa na symulatorze, a nie na rzeczywistym układzie kwantowym, wyniki sugerują, że metody inspirowane kwantowo mogą dodawać użyteczne niuanse do istniejących pipeline’ów uczenia maszynowego nawet na wczesnym etapie rozwoju.
Od komputerowych kart wyników do przyszłych testów laboratoryjnych
Po walidacji modeli toksyczności autorzy stosują pełny pipeline Q-CaDD do przefiltrowanej biblioteki skoncentrowanej na EGFR. Unikają twardej decyzji tak/nie dotyczącej toksyczności, zamiast tego zachowując ciągłe oceny ryzyka i łącząc je z oszacowaniami siły wiązania z dokowania. To generuje listę priorytetową kandydatów, z których niektóre wydają się wiązać z EGFR silniej niż lek referencyjny, przy jednoczesnym niskim przewidywanym poziomie toksyczności. Te molekuły nie są przedstawiane jako nowe leki; są wskazywane jako prowadzenia (leads), które zasługują na testy laboratoryjne. Główne przesłanie badania dla niespecjalistów nie brzmi, że komputery kwantowe już zrewolucjonizowały odkrywanie leków, lecz że starannie zaprojektowane hybrydy narzędzi klasycznych i inspirowanych kwantowo mogą już pomóc wyostrzyć poszukiwania, skierowując badaczy ku lepszym kandydatom szybciej, przy realistycznym podejściu do obecnych ograniczeń sprzętowych.
Cytowanie: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Słowa kluczowe: kwantowe odkrywanie leków, inhibitory EGFR, toksyczność uczenie maszynowe, wirtualne przesiewanie, niedrobnokomórkowy rak płuca