Clear Sky Science · de
Q-CaDD: Beschleunigung in silico-Methoden mit Quantencomputing und maschinellem Lernen für den Epidermalen Wachstumsfaktor-Rezeptor
Warum neue Computerwerkzeuge für künftige Krebsmedikamente wichtig sind
Neue Arzneimittel zu entwerfen ist ein bisschen so, als würde man eine Nadel in einem Heuhaufen möglicher Moleküle suchen. Bei Krebserkrankungen, die durch ein Protein namens Epidermaler Wachstumsfaktor-Rezeptor (EGFR) angetrieben werden, müssen Forscher Verbindungen finden, die dieses Protein fest binden und zugleich für Patientinnen und Patienten sicher bleiben. Dieses Paper stellt Q-CaDD vor, ein computerbasiertes Rahmenwerk, das heutiges maschinelles Lernen mit aufkommenden Ideen aus dem Quantencomputing verbindet, um Hunderttausende potenzieller Moleküle effizienter zu durchsieben und diejenigen zu markieren, die sicherere, wirksamere Wirkstoffkandidaten werden könnten.
Vom krebsassoziierten Protein zum digitalen Suchproblem
EGFR sitzt auf der Zelloberfläche und hilft zu steuern, wie Zellen wachsen und sich teilen. Wenn er fehlfunktioniert, wie das häufig beim nicht-kleinzelligen Lungenkrebs der Fall ist, können sich Zellen ungezügelt vermehren. Es gibt bereits Wirkstoffe, die EGFR blockieren, doch Tumoren können resistent werden und nicht jede Patientin bzw. jeder Patient spricht gut an. Statt neue Verbindungen einzeln im Labor zu testen, nutzt Q-CaDD Computersimulationen, um den chemischen Raum in großem Maßstab zu untersuchen und Moleküle zu finden, die sowohl an EGFR andocken als auch Anzeichen niedriger Toxizität zeigen. Dieser Ansatz zielt darauf ab, die frühen Schritte der Wirkstoffsuche schneller, günstiger und zielgerichteter zu machen.
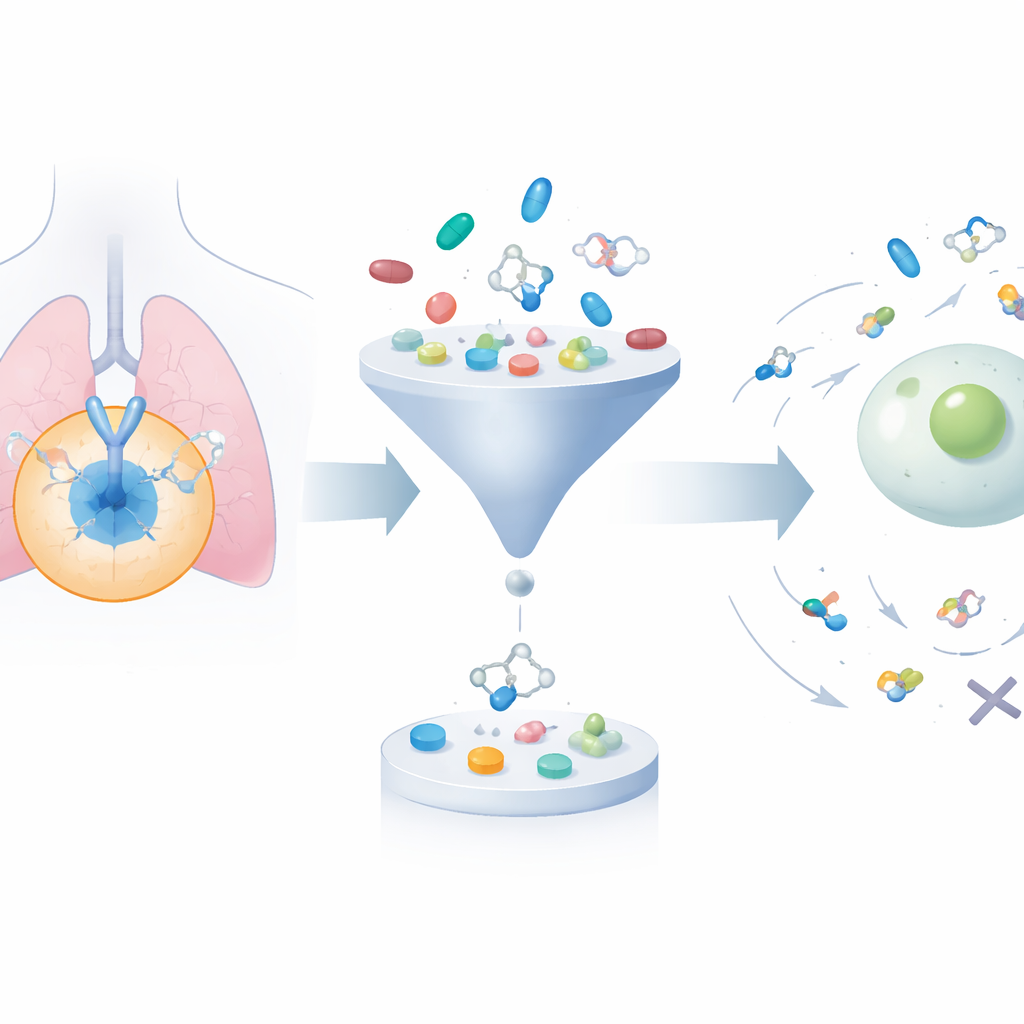
Aufbau und Beschneidung einer riesigen Molekülbibliothek
Das Rahmenwerk beginnt mit der Sammlung von etwa 24.000 bekannten EGFR-blockierenden Molekülen aus öffentlichen Datenbanken. Anschließend verwendet es einen generativen Algorithmus, um deren Strukturen systematisch zu verändern und so rund 200.000 verwandte Kandidaten zu erzeugen. Zwei etablierte Filter zur „Drug-Likeness“ werden angewandt, um Verbindungen auszusortieren, die zu sperrig, zu fettlöslich oder anderweitig unwahrscheinlich sind, sich im Körper gut zu verhalten, wodurch die Menge auf weniger als 50.000 reduziert wird. Danach platziert ein Docking-Programm jedes Molekül virtuell in die dreidimensionale Tasche auf EGFR, in der reale Wirkstoffe binden würden, und schätzt, wie stark sich jedes anlagern könnte. So konzentriert sich die Auswahl auf Verbindungen, die sowohl chemisch plausibel sind als auch voraussichtlich gut mit dem Ziel interagieren.
Computern toxische Warnsignale beibringen
Die Bindung an EGFR ist nur die halbe Geschichte; ein vielversprechendes Molekül darf auch kein gesundes Gewebe schädigen. Zur Abschätzung der Toxizität greift die Studie auf einen großen öffentlichen Datensatz namens Tox21 zurück, der dokumentiert, wie über 10.000 Chemikalien verschiedene zelluläre Signalwege beeinflussen. Die Autoren konzentrieren sich auf einen Weg, der mit dem Androgenrezeptor verknüpft ist, ausgewählt, weil er gut annotiert und biologisch relevant für mehrere Krebsarten ist. Jedes Tox21-Molekül wird in einen numerischen Fingerprint übersetzt, der seine strukturellen Merkmale und Ähnlichkeiten zu anderen Chemikalien erfasst. Diese Fingerprints speisen mehrere Vorhersagemodelle, darunter neuronale Netze, Entscheidungsbäume, eine klassische Support-Vektor-Maschine und eine quanteninspirierte Support-Vektor-Maschine, die einen einfachen Quanten-Schaltkreis nutzt, um Verbindungen in einem anderen mathematischen Raum zu vergleichen.
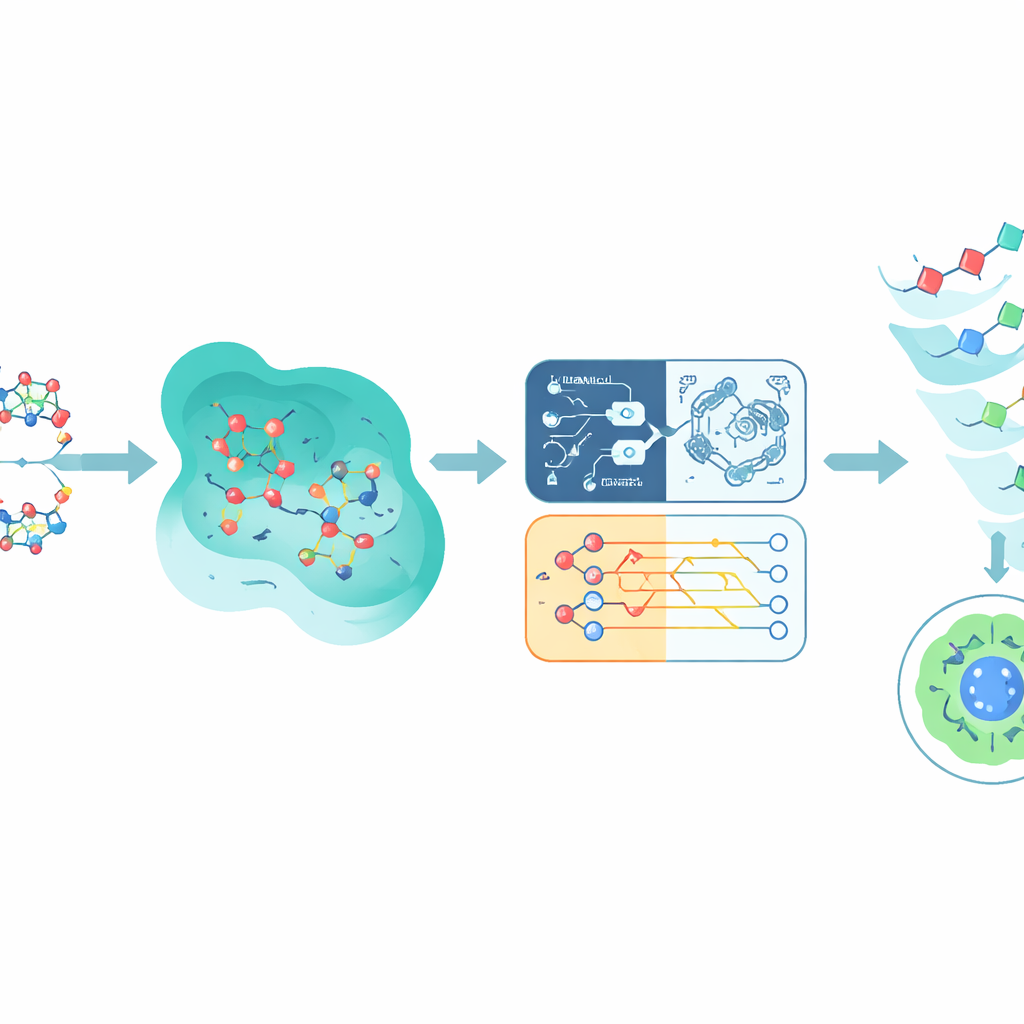
Verschmelzung quantenbasierter und klassischer Vorhersagen
Statt auf ein einzelnes Modell zu setzen, kombiniert Q-CaDD die Ausgaben aller vier Modelle zu einem Ensemble, gewichtet das neuronale Netz am stärksten, bezieht aber weiterhin das schwächere, doch unterschiedliche Signal des Quantenmodells mit ein. Beim Testen an bislang ungesehenen Tox21-Daten übertrifft dieser gemischte Ansatz jedes einzelne Modell darin, toxischere von weniger toxischen Verbindungen zu unterscheiden, gemessen an einer Standardbewertungsgröße, der Fläche unter der ROC-Kurve. Obwohl die Verbesserung modest ist und der Quantenanteil noch auf einem Simulator statt auf einem echten Quantenchip läuft, deuten die Ergebnisse darauf hin, dass quanteninspirierte Methoden bereits jetzt nützliche Nuancen zu bestehenden maschinellen Lernpipelines beitragen können — selbst in frühen Stadien.
Von Computer-Scorekarten zu künftigen Labortests
Nach der Validierung der Toxizitätsmodelle wenden die Autoren die vollständige Q-CaDD-Pipeline auf die gefilterte, EGFR-fokussierte Bibliothek an. Sie vermeiden eine harte Ja-/Nein-Entscheidung zur Toxizität, behalten stattdessen kontinuierliche Risikoscores bei und kombinieren diese mit den Docking-Schätzungen zur Bindungsstärke. Das Ergebnis ist eine Prioritätenliste von Molekülkandidaten, von denen einige voraussichtlich stärker an EGFR binden als ein Referenzmedikament und zugleich eine niedrige vorhergesagte Toxizität aufweisen. Diese Moleküle werden nicht als neue Medikamente beansprucht; sie werden als Leads markiert, die labortechnisch überprüft werden sollten. Die wichtigste Erkenntnis für Nicht-Spezialisten ist nicht, dass Quantencomputer die Wirkstoffforschung bereits revolutioniert haben, sondern dass sorgfältig gestaltete Hybride aus klassischen und quanteninspirierten Werkzeugen bereits jetzt die Suche schärfen können, Forschende schneller zu besseren Wirkstoffkandidaten führen und zugleich realistisch in Bezug auf die aktuellen Hardwaregrenzen bleiben.
Zitation: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Schlüsselwörter: quantitative Wirkstoffentdeckung, EGFR-Inhibitoren, Toxizität maschinelles Lernen, virtuelles Screening, Nicht-kleinzelliger Lungenkrebs