Clear Sky Science · en
Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor
Why new computer tools matter for future cancer drugs
Designing new medicines is a bit like searching for a needle in a haystack of possible molecules. For cancers driven by a protein called the Epidermal Growth Factor Receptor (EGFR), researchers must find compounds that bind this protein tightly yet remain safe for patients. This paper introduces Q-CaDD, a computer-based framework that blends today’s machine learning with emerging quantum computing ideas to sift through hundreds of thousands of candidate molecules more efficiently and to flag those that might become safer, more effective drugs.
From a cancer-linked protein to a digital search problem
EGFR sits on the surface of cells and helps control how they grow and divide. When it malfunctions, as it often does in non-small cell lung cancer, cells can multiply uncontrollably. Drugs that block EGFR already exist, but cancers can become resistant, and not every patient responds well. Instead of testing new compounds one by one in the lab, Q-CaDD uses computer simulations to explore chemical space in bulk, looking for molecules that both latch onto EGFR and show signs of low toxicity. This approach aims to make the early steps of drug discovery faster, cheaper, and more guided.
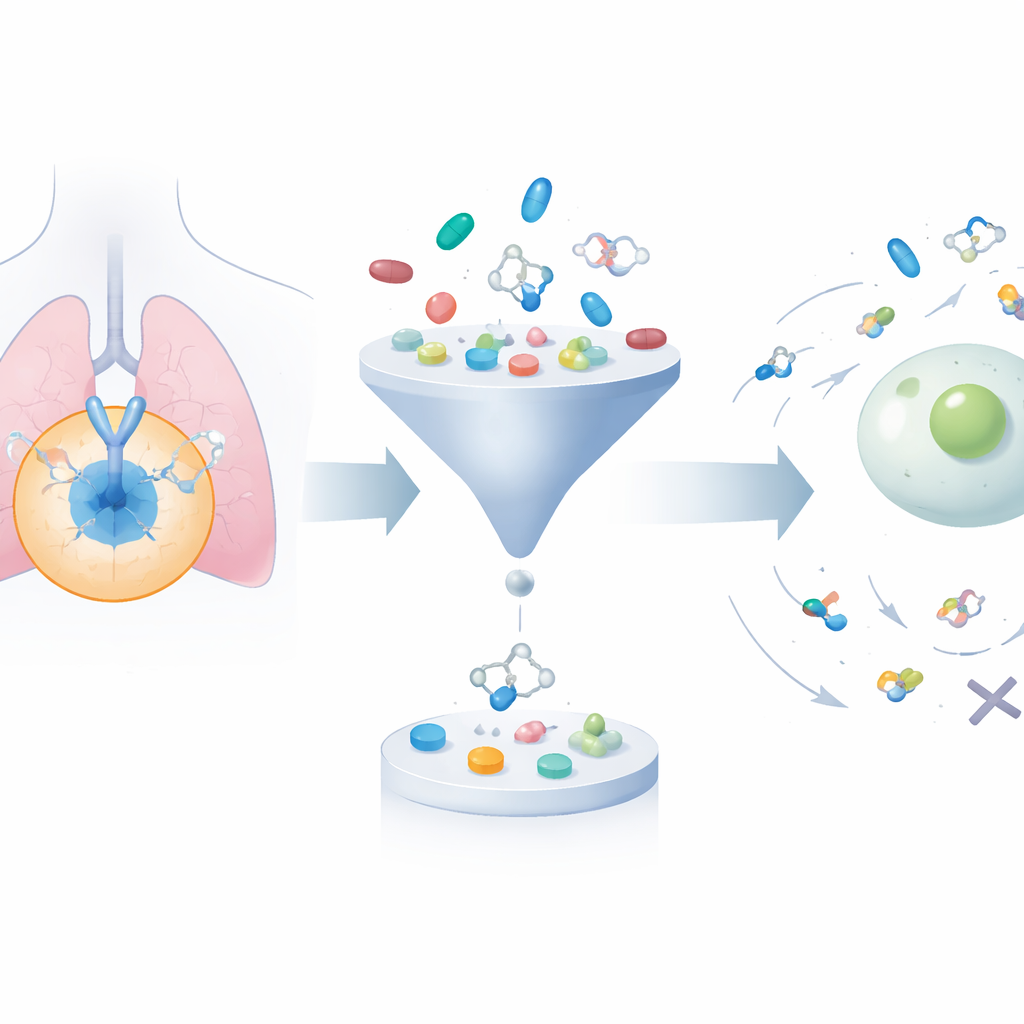
Growing and trimming a vast library of molecules
The framework begins by collecting about 24,000 known EGFR-blocking molecules from public databases. It then uses a generative algorithm to systematically tweak their structures, producing roughly 200,000 related candidates. Two well-established “drug-likeness” filters are applied to weed out compounds that are too bulky, too greasy, or otherwise unlikely to behave well in the body, cutting the set down to fewer than 50,000. Next, a docking program virtually fits each molecule into the three-dimensional pocket on EGFR where real drugs would bind, estimating how strongly each one might attach. This narrows attention to compounds that are both chemically reasonable and predicted to interact well with the target.
Teaching computers to recognize toxic warning signs
Binding to EGFR is only half the story; a promising compound must also avoid harming healthy tissues. To estimate toxicity, the study turns to a large public dataset called Tox21, which records how over 10,000 chemicals affect various cellular pathways. The authors focus on one pathway linked to the androgen receptor, chosen because it is well annotated and biologically relevant to several cancers. Each Tox21 molecule is translated into a numerical fingerprint that captures its structural features and similarities to other chemicals. These fingerprints feed several predictive models, including neural networks, decision trees, a traditional support vector machine, and a quantum-inspired support vector machine that uses a simple quantum circuit to compare compounds in a different mathematical space.
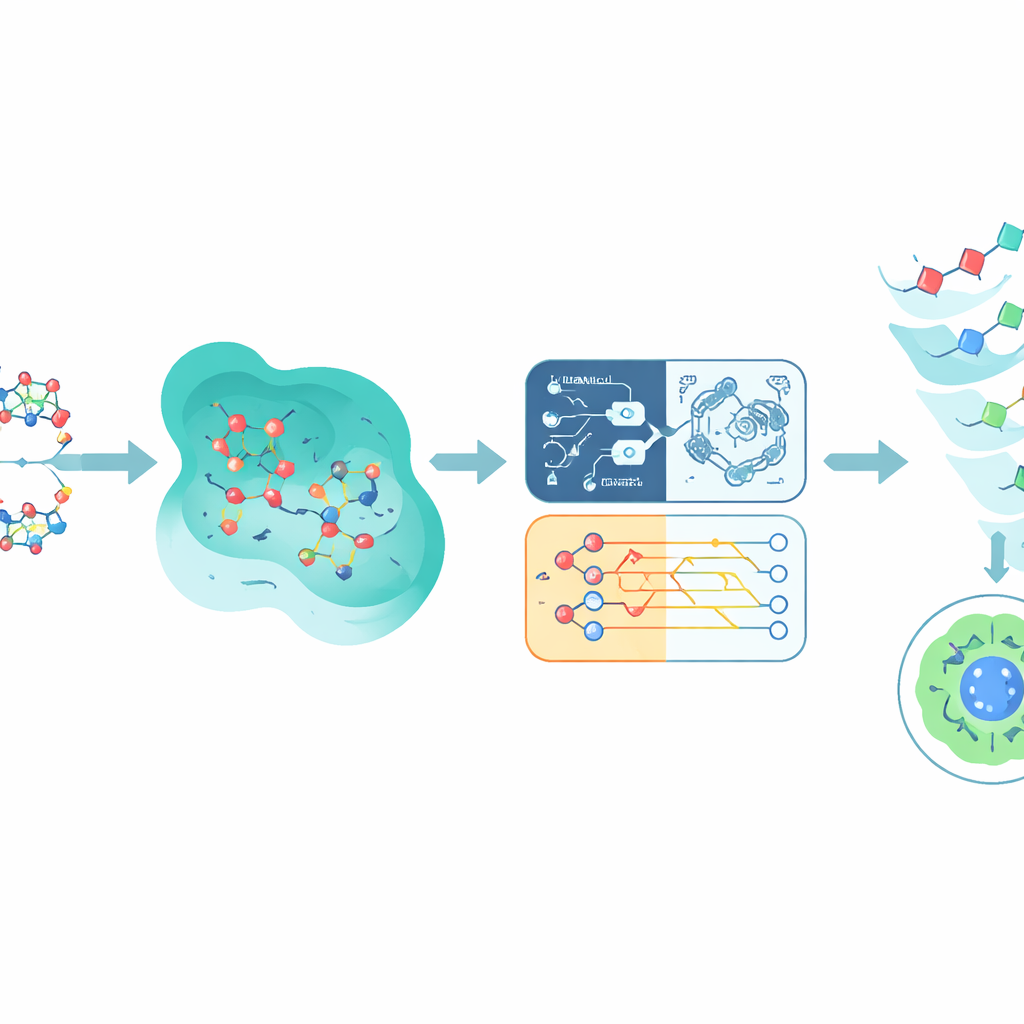
Blending quantum and classical predictions
Rather than betting on a single model, Q-CaDD combines the outputs of all four into an ensemble, giving the greatest weight to the neural network but still incorporating the weaker yet distinct signal from the quantum model. When tested on previously unseen Tox21 data, this blended approach outperforms any individual model at distinguishing more and less toxic compounds, as measured by a standard ranking score called the area under the ROC curve. Although the improvement is modest and the quantum part is still run on a simulator rather than a real quantum chip, the results suggest that quantum-inspired methods can add useful nuance to existing machine learning pipelines even in their early stages.
From computer scorecards to future lab tests
After validating the toxicity models, the authors apply Q-CaDD’s full pipeline to the filtered EGFR-focused library. They avoid making a hard yes-or-no call on toxicity, instead keeping continuous risk scores and combining them with docking estimates of binding strength. This produces a priority list of candidate molecules, some of which appear to bind EGFR more strongly than a reference drug while retaining low predicted toxicity. These molecules are not claimed as new medicines; they are flagged as leads that merit laboratory testing. The study’s main takeaway for non-specialists is not that quantum computers have already revolutionized drug discovery, but that carefully designed hybrids of classical and quantum-inspired tools can already help sharpen the search, pointing researchers toward better drug candidates faster while staying realistic about current hardware limits.
Citation: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Keywords: quantum drug discovery, EGFR inhibitors, machine learning toxicity, virtual screening, non-small cell lung cancer