Clear Sky Science · nl
Q-CaDD: versnelling van in silico-methoden met kwantumcomputing en machine learning voor de Epidermal growth factor receptor
Waarom nieuwe computertools belangrijk zijn voor toekomstige kankergeneesmiddelen
Het ontwerpen van nieuwe medicijnen is een beetje alsof je een naald zoekt in een hooiberg van mogelijke moleculen. Voor kankers die worden aangedreven door een eiwit genaamd de Epidermal Growth Factor Receptor (EGFR) moeten onderzoekers verbindingen vinden die sterk aan dit eiwit binden maar tegelijk veilig blijven voor patiënten. Dit artikel introduceert Q-CaDD, een computergebaseerd kader dat hedendaagse machine learning combineert met opkomende ideeën uit de kwantumcomputing om efficiënter door honderden duizenden kandidaat-moleculen te zoeken en die te markeren die mogelijk veiliger en effectiever geneesmiddelen kunnen worden.
Van een aan kanker gelinkt eiwit naar een digitaal zoekprobleem
EGFR bevindt zich op het celoppervlak en helpt de groei en deling van cellen te reguleren. Wanneer het niet goed functioneert, zoals vaak het geval is bij niet-kleincellige longkanker, kunnen cellen zich ongecontroleerd vermenigvuldigen. Er bestaan al geneesmiddelen die EGFR blokkeren, maar kankers kunnen resistent raken en niet elke patiënt reageert goed. In plaats van nieuwe verbindingen één voor één in het laboratorium te testen, gebruikt Q-CaDD computersimulaties om in bulk de chemische ruimte te verkennen, op zoek naar moleculen die zowel aan EGFR hechten als tekenen van lage toxiciteit vertonen. Deze aanpak heeft tot doel de vroege stappen van geneesmiddelenontdekking sneller, goedkoper en beter gestuurd te maken.
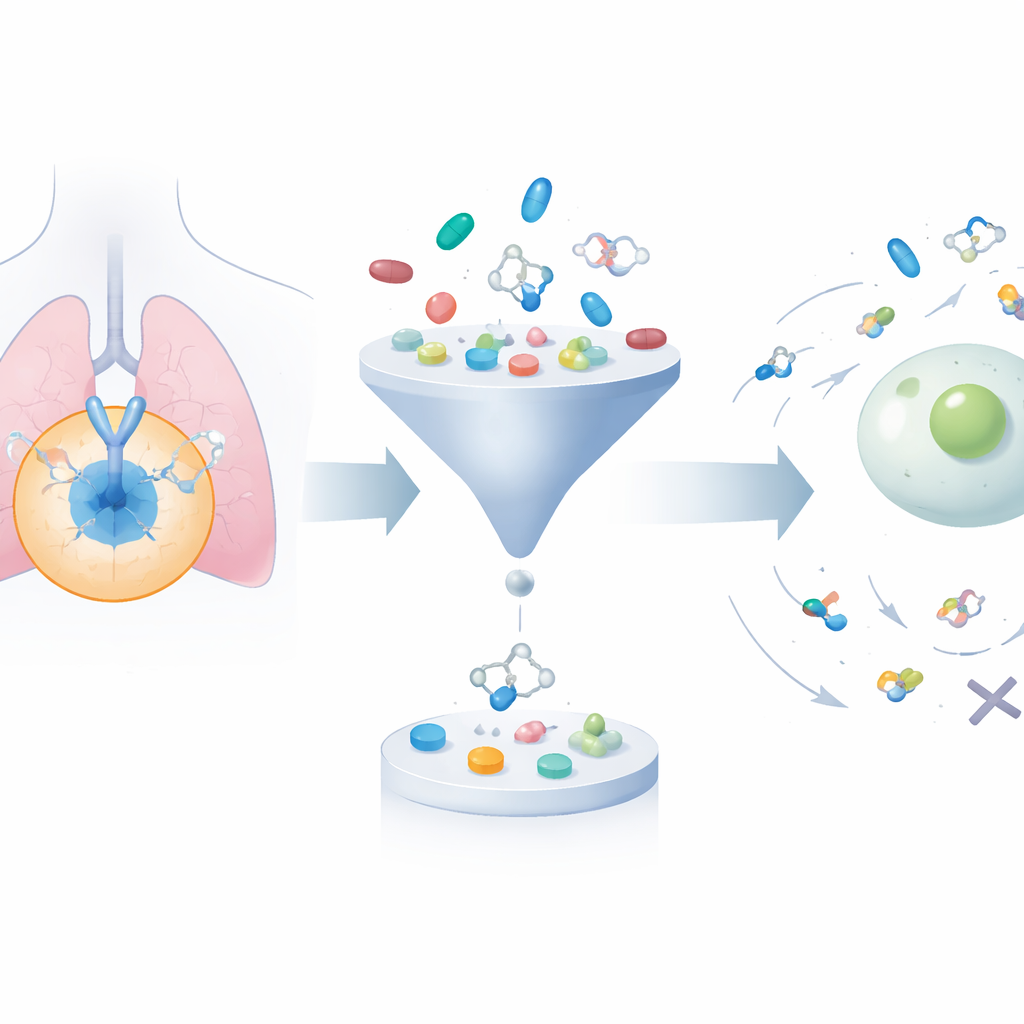
Een enorme bibliotheek van moleculen uitbreiden en verfijnen
Het kader begint met het verzamelen van ongeveer 24.000 bekende EGFR-blokkerende moleculen uit openbare databases. Vervolgens gebruikt het een generatief algoritme om hun structuren systematisch aan te passen, waardoor ongeveer 200.000 verwante kandidaten ontstaan. Twee goed gevestigde filters voor “drug-likeness” worden toegepast om verbindingen te weren die te omvangrijk, te vet of anderszins onwaarschijnlijk zijn om goed in het lichaam te functioneren, waardoor de set wordt teruggebracht tot minder dan 50.000. Daarna past een dockingprogramma elk molecuul virtueel in het driedimensionale pocket op EGFR waar echte geneesmiddelen zouden binden en schat het in hoe sterk elk ervan zou kunnen hechten. Dit beperkt de aandacht tot verbindingen die zowel chemisch redelijk zijn als naar verwachting goed met het doelwit interageren.
Computers leren toxische waarschuwingssignalen te herkennen
Aan EGFR binden is slechts de helft van het verhaal; een veelbelovende verbinding moet ook vermijden gezonde weefsels te schaden. Om toxiciteit te schatten, maakt de studie gebruik van een grote openbare dataset genaamd Tox21, die vastlegt hoe meer dan 10.000 chemicaliën verschillende cellulaire routes beïnvloeden. De auteurs concentreren zich op één route die verband houdt met de androgenenreceptor, gekozen omdat deze goed geannoteerd en biologisch relevant is voor meerdere kankers. Elk Tox21-molecuul wordt omgezet in een numerieke vingerafdruk die zijn structurele kenmerken en overeenkomsten met andere chemicaliën vastlegt. Deze vingerafdrukken voeden verschillende voorspellende modellen, waaronder neurale netwerken, beslisbomen, een traditionele support vector machine en een kwantuminspireerde support vector machine die een eenvoudige kwantumcircuit gebruikt om verbindingen in een andere wiskundige ruimte te vergelijken.
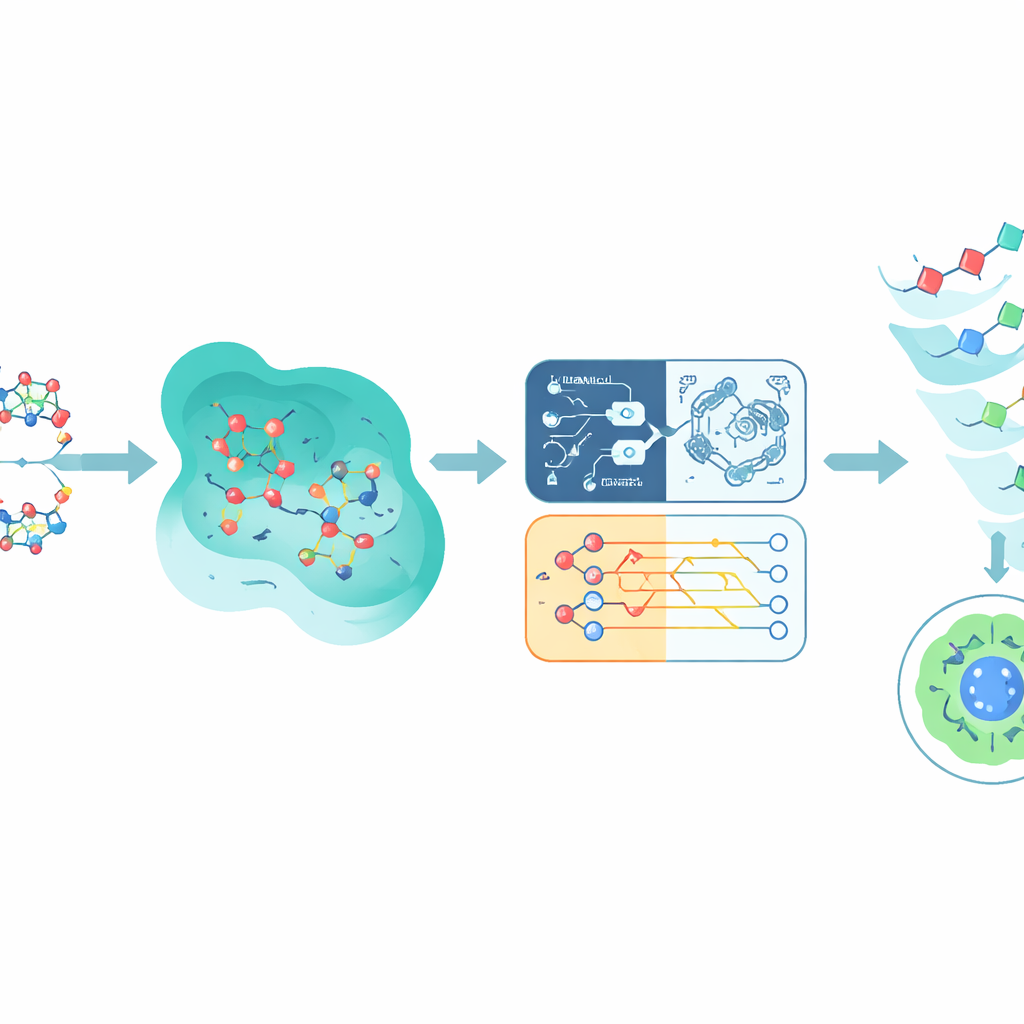
Kwantum- en klassieke voorspellingen combineren
In plaats van te wedden op één model combineert Q-CaDD de outputs van alle vier in een ensemble, waarbij het grootste gewicht wordt gegeven aan het neurale netwerk maar ook het zwakkere doch onderscheidende signaal van het kwantummodel wordt opgenomen. Getest op eerder niet geziene Tox21-gegevens, presteert deze geblende aanpak beter dan elk individueel model in het onderscheiden van meer en minder toxische verbindingen, gemeten met een standaard rangschikkingsscore genaamd de area under the ROC curve. Hoewel de verbetering bescheiden is en het kwantumdeel nog op een simulator draait in plaats van op een echte kwantumchip, suggereren de resultaten dat kwantuminspireerde methoden nuttige nuance kunnen toevoegen aan bestaande machine learning-pijplijnen, zelfs in hun vroegste fasen.
Van computerrapporten naar toekomstige labtests
Na het valideren van de toxiciteitsmodellen passen de auteurs de volledige Q-CaDD-pijplijn toe op de gefilterde EGFR-gerichte bibliotheek. Ze vermijden een harde ja- of nee-beslissing over toxiciteit en behouden in plaats daarvan continue risico-scores die gecombineerd worden met docking-schattingen van bindingssterkte. Dit levert een prioriteitenlijst van kandidaat-moleculen op, waarvan sommige lijken sterker aan EGFR te binden dan een referentiegeneesmiddel terwijl ze lage voorspelde toxiciteit behouden. Deze moleculen worden niet opgevoerd als nieuwe geneesmiddelen; ze worden aangeduid als leads die laboratoriumtesten verdienen. De belangrijkste conclusie van de studie voor niet-specialisten is niet dat kwantumcomputers de geneesmiddelenontdekking al hebben gerevolutioneerd, maar dat zorgvuldig ontworpen hybriden van klassieke en kwantuminspireerde hulpmiddelen nu al kunnen helpen de zoektocht te verscherpen en onderzoekers sneller naar betere geneesmiddelkandidaten te wijzen, terwijl ze realistisch blijven over de huidige hardwarelimieten.
Bronvermelding: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Trefwoorden: kwantum geneesmiddelenontdekking, EGFR-remmers, machine learning toxiciteit, virtuele screening, niet-kleincellige longkanker