Clear Sky Science · fr
Q-CaDD : accélérer les méthodologies in silico avec le calcul quantique et l'apprentissage automatique pour le récepteur du facteur de croissance épidermique
Pourquoi de nouveaux outils informatiques comptent pour les médicaments anticancéreux futurs
Concevoir de nouveaux médicaments revient un peu à chercher une aiguille dans une meule de foin de molécules possibles. Pour les cancers pilotés par une protéine appelée récepteur du facteur de croissance épidermique (EGFR), les chercheurs doivent trouver des composés qui se lient fortement à cette protéine tout en restant sûrs pour les patients. Cet article présente Q-CaDD, un cadre informatique qui mêle l'apprentissage automatique actuel à des idées émergentes du calcul quantique pour trier des centaines de milliers de molécules candidates plus efficacement et repérer celles susceptibles de devenir des médicaments plus sûrs et plus efficaces.
D'une protéine liée au cancer à un problème de recherche numérique
EGFR se situe à la surface des cellules et aide à contrôler leur croissance et leur division. Lorsqu'il dysfonctionne, comme c'est souvent le cas dans le cancer du poumon non à petites cellules, les cellules peuvent se multiplier de façon incontrôlée. Des médicaments qui bloquent EGFR existent déjà, mais les cancers peuvent devenir résistants et tous les patients ne répondent pas bien. Plutôt que de tester de nouveaux composés un par un au laboratoire, Q-CaDD utilise des simulations informatiques pour explorer l'espace chimique en masse, à la recherche de molécules qui se fixent à EGFR et présentent des signes de faible toxicité. Cette approche vise à rendre les premières étapes de la découverte de médicaments plus rapides, moins coûteuses et mieux guidées.
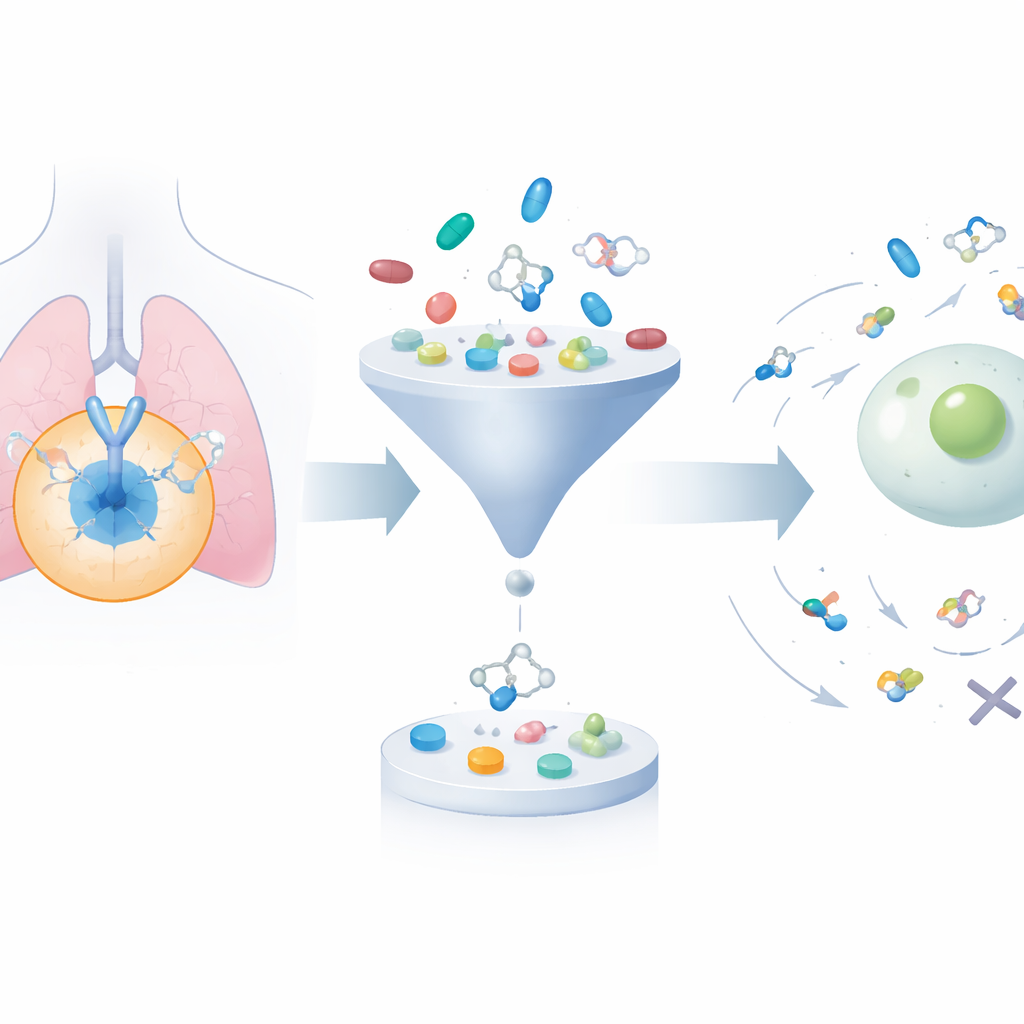
Faire croître puis tailler une vaste bibliothèque de molécules
Le cadre commence par rassembler environ 24 000 molécules connues pour bloquer EGFR à partir de bases de données publiques. Il utilise ensuite un algorithme génératif pour modifier systématiquement leurs structures, produisant environ 200 000 candidats apparentés. Deux filtres bien établis de « drug-likeness » sont appliqués pour éliminer les composés trop volumineux, trop lipidiques ou autrement peu susceptibles de bien se comporter dans l'organisme, réduisant l'ensemble à moins de 50 000. Ensuite, un programme de docking ajuste virtuellement chaque molécule dans la poche tridimensionnelle d'EGFR où les médicaments réels se lieraient, estimant la force probable de chaque liaison. Cela concentre l'attention sur des composés à la fois chimiquement raisonnables et prédits pour interagir efficacement avec la cible.
Apprendre aux ordinateurs à reconnaître les signaux de danger toxique
La liaison à EGFR ne constitue que la moitié de l'histoire ; un composé prometteur doit aussi éviter d'endommager les tissus sains. Pour estimer la toxicité, l'étude s'appuie sur un large jeu de données public appelé Tox21, qui enregistre comment plus de 10 000 produits chimiques affectent diverses voies cellulaires. Les auteurs se concentrent sur une voie liée au récepteur aux androgènes, choisie parce qu'elle est bien annotée et biologiquement pertinente pour plusieurs cancers. Chaque molécule de Tox21 est traduite en une empreinte numérique qui capture ses caractéristiques structurelles et ses similarités avec d'autres produits chimiques. Ces empreintes alimentent plusieurs modèles prédictifs, y compris des réseaux de neurones, des arbres de décision, une machine à vecteurs de support traditionnelle et une machine à vecteurs de support inspirée du quantique qui utilise un circuit quantique simple pour comparer les composés dans un espace mathématique différent.
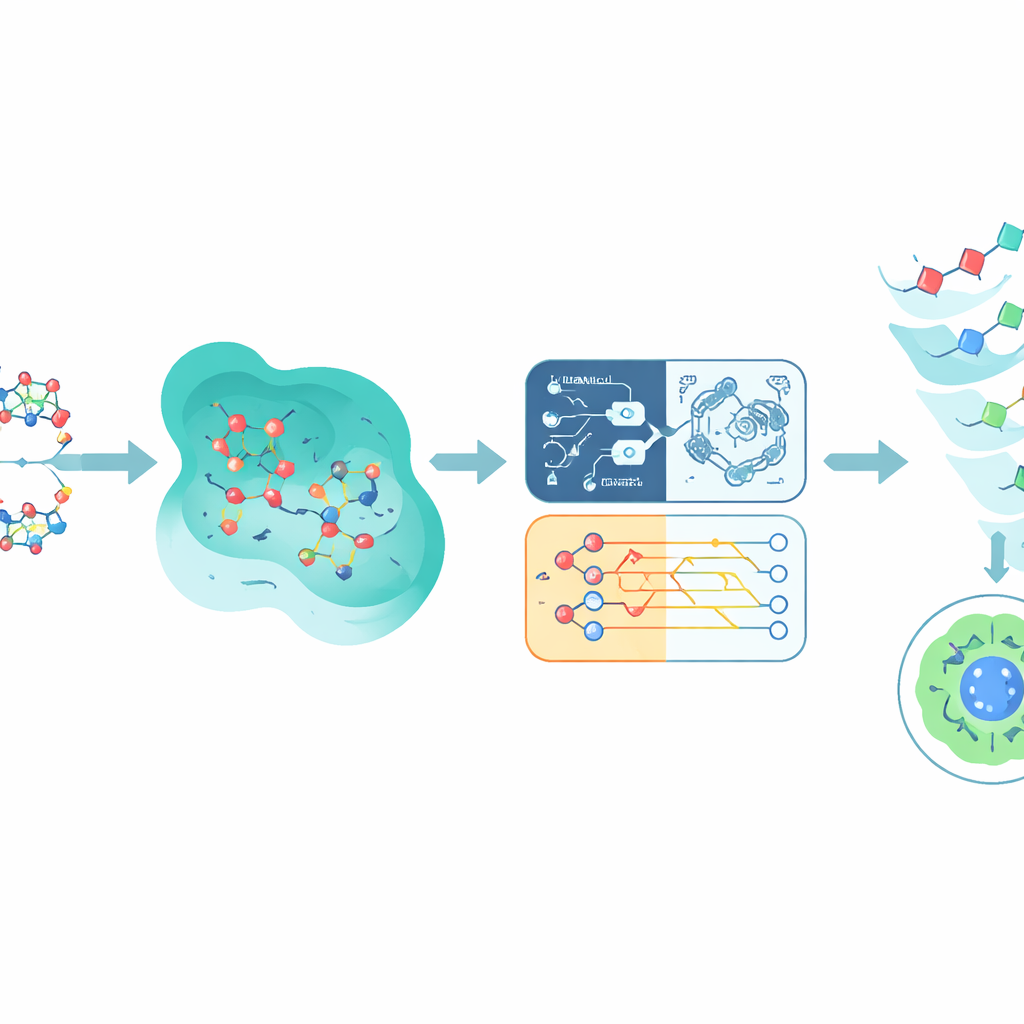
Mêler prédictions quantiques et classiques
Plutôt que de parier sur un seul modèle, Q-CaDD combine les sorties des quatre modèles dans un ensemble, en donnant le plus de poids au réseau de neurones tout en incorporant le signal plus faible mais distinct du modèle quantique. Testée sur des données Tox21 non vues auparavant, cette approche combinée surpasse chaque modèle individuel pour distinguer les composés plus ou moins toxiques, mesurée par un score de classement standard appelé aire sous la courbe ROC. Bien que l'amélioration soit modeste et que la partie quantique soit encore exécutée sur un simulateur plutôt que sur une puce quantique réelle, les résultats suggèrent que les méthodes inspirées du quantique peuvent apporter une nuance utile aux pipelines d'apprentissage automatique existants, même à leurs débuts.
Des bulletins informatiques aux futurs tests en laboratoire
Après avoir validé les modèles de toxicité, les auteurs appliquent le pipeline complet de Q-CaDD à la bibliothèque filtrée axée sur l'EGFR. Ils évitent un jugement catégorique oui/non sur la toxicité, préférant conserver des scores de risque continus et les combiner avec les estimations de liaison issues du docking. Cela produit une liste de priorités de molécules candidates, dont certaines semblent se lier à EGFR plus fortement qu'un médicament de référence tout en conservant une faible toxicité prédite. Ces molécules ne sont pas présentées comme de nouveaux médicaments ; elles sont signalées comme des candidats qui méritent des tests en laboratoire. Le principal enseignement de l'étude pour un public non spécialiste n'est pas que les ordinateurs quantiques ont déjà révolutionné la découverte de médicaments, mais que des hybrides soigneusement conçus d'outils classiques et inspirés du quantique peuvent déjà aider à affiner la recherche, orientant plus rapidement les chercheurs vers de meilleurs candidats tout en restant réalistes quant aux limites matérielles actuelles.
Citation: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Mots-clés: découverte de médicaments quantique, inhibiteurs EGFR, toxicité apprentissage automatique, dépistage virtuel, cancer du poumon non à petites cellules