Clear Sky Science · pt
Q-CaDD: acelerando metodologias in silico com computação quântica e aprendizado de máquina para o receptor do fator de crescimento epidérmico
Por que novas ferramentas computacionais importam para os medicamentos do futuro
Projetar novos medicamentos é um pouco como procurar uma agulha num palheiro de moléculas possíveis. Para cânceres impulsionados por uma proteína chamada Receptor do Fator de Crescimento Epidérmico (EGFR), os pesquisadores precisam encontrar compostos que se liguem firmemente a essa proteína e, ao mesmo tempo, sejam seguros para os pacientes. Este artigo apresenta o Q-CaDD, uma estrutura baseada em computador que combina o aprendizado de máquina atual com ideias emergentes de computação quântica para vasculhar centenas de milhares de moléculas candidatas de forma mais eficiente e identificar aquelas que podem se tornar medicamentos mais seguros e eficazes.
De uma proteína ligada ao câncer a um problema de busca digital
O EGFR fica na superfície das células e ajuda a controlar como elas crescem e se dividem. Quando ele funciona mal, como frequentemente acontece no câncer de pulmão de não pequenas células, as células podem se multiplicar descontroladamente. Já existem drogas que bloqueiam o EGFR, mas os cânceres podem desenvolver resistência, e nem todo paciente responde bem. Em vez de testar novos compostos um a um no laboratório, o Q-CaDD usa simulações computacionais para explorar o espaço químico em massa, procurando moléculas que tanto se prendam ao EGFR quanto mostrem sinais de baixa toxicidade. Essa abordagem visa tornar as etapas iniciais da descoberta de fármacos mais rápidas, mais baratas e mais direcionadas.
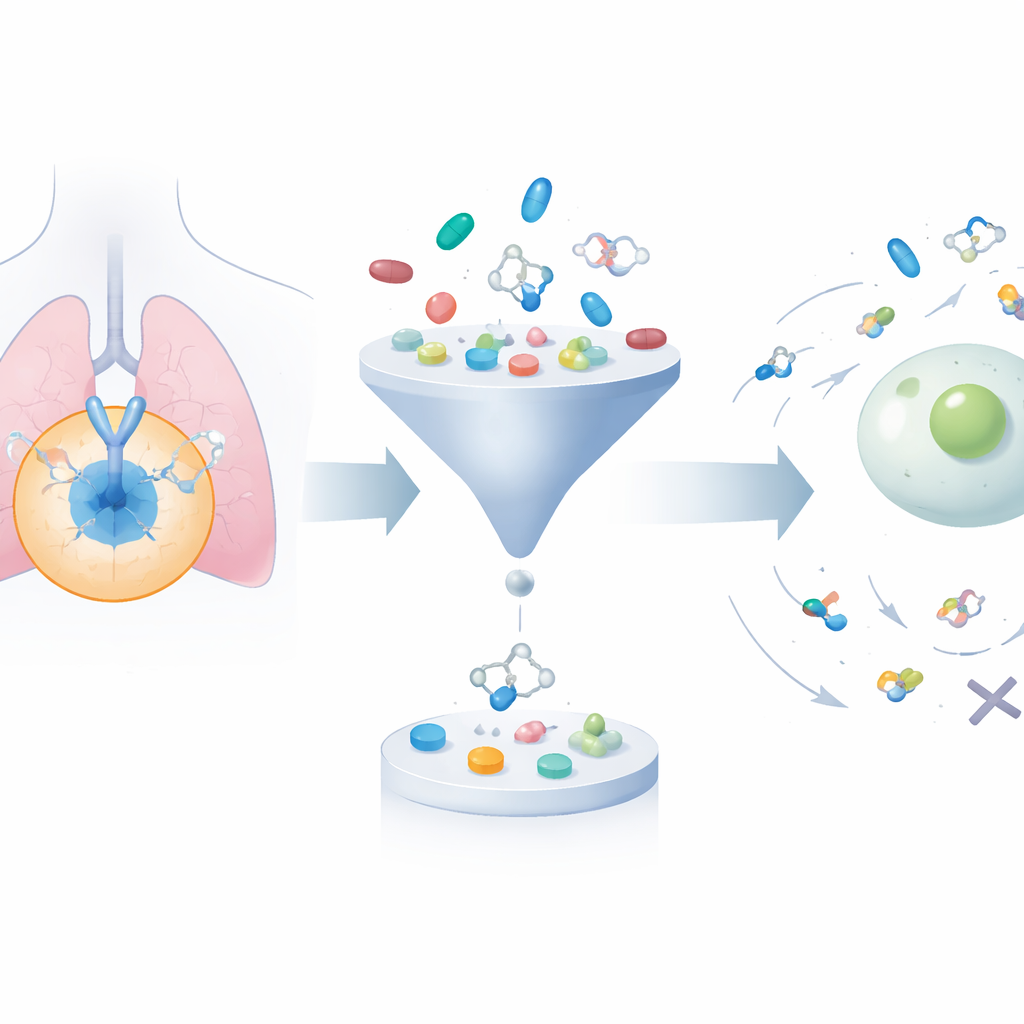
Crescendo e aparando uma vasta biblioteca de moléculas
A estrutura começa coletando cerca de 24.000 moléculas conhecidas por bloquear o EGFR de bases de dados públicas. Em seguida, usa um algoritmo generativo para ajustar sistematicamente suas estruturas, produzindo aproximadamente 200.000 candidatos relacionados. Dois filtros bem estabelecidos de “semblante de fármaco” são aplicados para eliminar compostos muito volumosos, muito lipofílicos ou que, de outra forma, provavelmente não se comportariam bem no organismo, reduzindo o conjunto para menos de 50.000. Depois, um programa de docking encaixa virtualmente cada molécula no bolso tridimensional do EGFR onde drogas reais se ligariam, estimando quão fortemente cada uma poderia se prender. Isso concentra a atenção em compostos que são ao mesmo tempo quimicamente razoáveis e previstos para interagir bem com o alvo.
Ensinando computadores a reconhecer sinais de alerta de toxicidade
Ligar-se ao EGFR é apenas metade da história; um composto promissor também deve evitar prejudicar tecidos saudáveis. Para estimar a toxicidade, o estudo recorre a um grande conjunto de dados público chamado Tox21, que registra como mais de 10.000 químicos afetam várias vias celulares. Os autores se concentram em uma via ligada ao receptor de andrógeno, escolhida por ser bem anotada e biologicamente relevante para vários cânceres. Cada molécula do Tox21 é traduzida em uma impressão digital numérica que captura suas características estruturais e semelhanças com outros químicos. Essas impressões alimentam vários modelos preditivos, incluindo redes neurais, árvores de decisão, uma máquina de vetores de suporte tradicional e uma máquina de vetores de suporte inspirada em computação quântica que usa um circuito quântico simples para comparar compostos em um espaço matemático diferente.
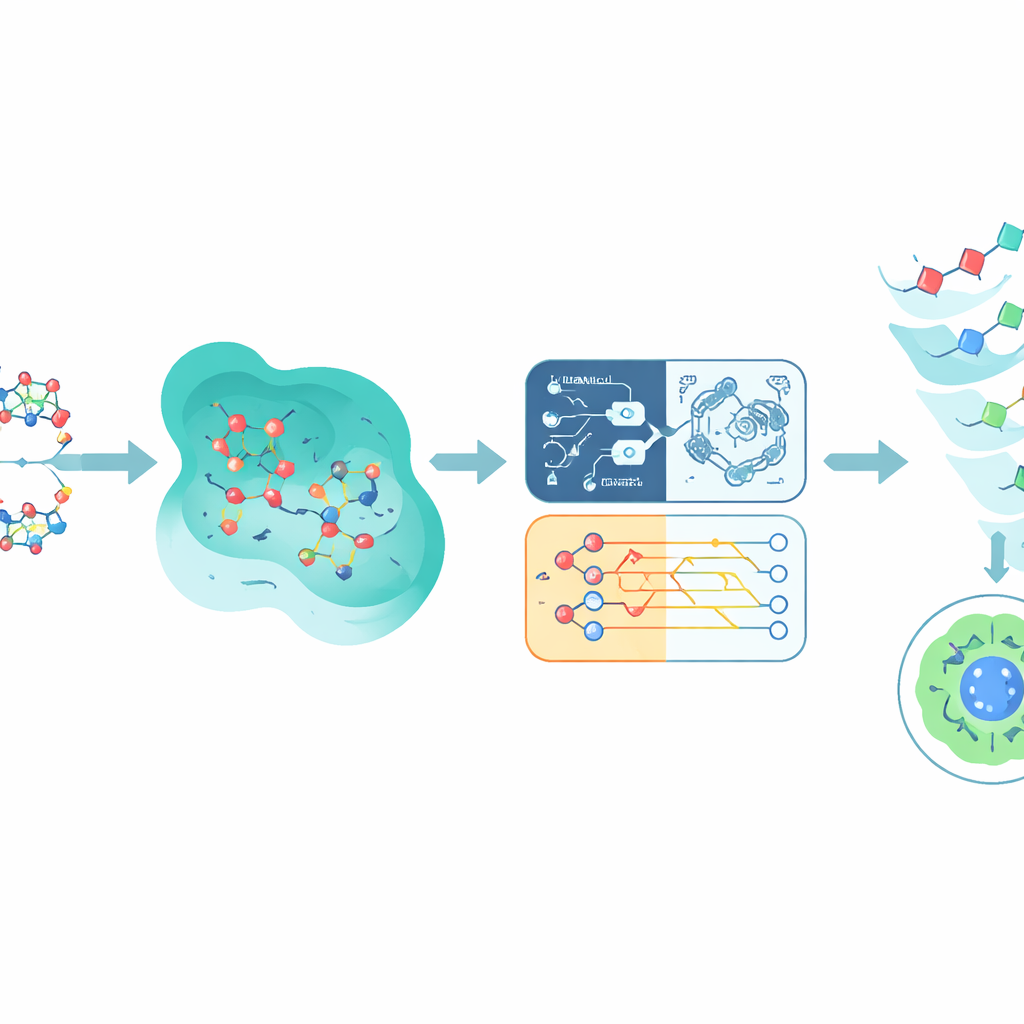
Misturando previsões quânticas e clássicas
Em vez de apostar em um único modelo, o Q-CaDD combina as saídas dos quatro em um conjunto, dando maior peso à rede neural, mas ainda incorporando o sinal mais fraco porém distinto do modelo quântico. Quando testada em dados Tox21 não vistos anteriormente, essa abordagem combinada supera qualquer modelo individual na distinção entre compostos mais e menos tóxicos, medida por uma pontuação padrão de ordenação chamada área sob a curva ROC. Embora a melhora seja modesta e a parte quântica ainda seja executada em um simulador em vez de um chip quântico real, os resultados sugerem que métodos inspirados em computação quântica podem acrescentar nuances úteis aos pipelines de aprendizado de máquina existentes, mesmo em estágios iniciais.
De boletins computacionais a testes laboratoriais futuros
Após validar os modelos de toxicidade, os autores aplicam o pipeline completo do Q-CaDD à biblioteca filtrada focada em EGFR. Eles evitam fazer um juízo binário de sim ou não sobre toxicidade, mantendo em vez disso escores contínuos de risco e combinando-os com estimativas de docking da força de ligação. Isso produz uma lista prioritária de moléculas candidatas, algumas das quais aparentam se ligar ao EGFR mais fortemente do que um fármaco de referência, mantendo baixa toxicidade prevista. Essas moléculas não são apresentadas como novos medicamentos; elas são sinalizadas como candidatos que merecem testes laboratoriais. A principal conclusão do estudo para não especialistas não é que computadores quânticos já revolucionaram a descoberta de fármacos, mas que híbridos bem desenhados de ferramentas clássicas e inspiradas em quântica já podem ajudar a afinar a busca, apontando os pesquisadores para candidatos melhores mais rapidamente, mantendo realismo quanto aos limites do hardware atual.
Citação: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Palavras-chave: descoberta de fármacos quântica, inibidores de EGFR, toxicidade por aprendizado de máquina, triagem virtual, câncer de pulmão de não pequenas células