Clear Sky Science · es
Q-CaDD: acelerando metodologías in silico con computación cuántica y aprendizaje automático para el receptor del factor de crecimiento epidérmico
Por qué importan las nuevas herramientas informáticas para los fármacos del futuro
Diseñar nuevos medicamentos es algo parecido a buscar una aguja en un pajar de moléculas posibles. Para los cánceres impulsados por una proteína llamada receptor del factor de crecimiento epidérmico (EGFR), los investigadores deben encontrar compuestos que se unan con firmeza a esta proteína pero que sigan siendo seguros para los pacientes. Este trabajo presenta Q-CaDD, un marco computacional que mezcla el aprendizaje automático actual con ideas emergentes de la computación cuántica para cribar cientos de miles de moléculas candidatas de manera más eficiente y señalar las que podrían convertirse en fármacos más seguros y efectivos.
De una proteína asociada al cáncer a un problema de búsqueda digital
EGFR se encuentra en la superficie de las células y ayuda a controlar cómo crecen y se dividen. Cuando funciona mal, como ocurre con frecuencia en el cáncer de pulmón no microcítico, las células pueden multiplicarse sin control. Ya existen fármacos que bloquean EGFR, pero los tumores pueden volverse resistentes y no todos los pacientes responden bien. En lugar de probar compuestos nuevos uno por uno en el laboratorio, Q-CaDD utiliza simulaciones por ordenador para explorar el espacio químico a gran escala, buscando moléculas que tanto se fijen a EGFR como presenten indicios de baja toxicidad. Este enfoque pretende hacer los primeros pasos del descubrimiento de fármacos más rápidos, más baratos y más dirigidos.
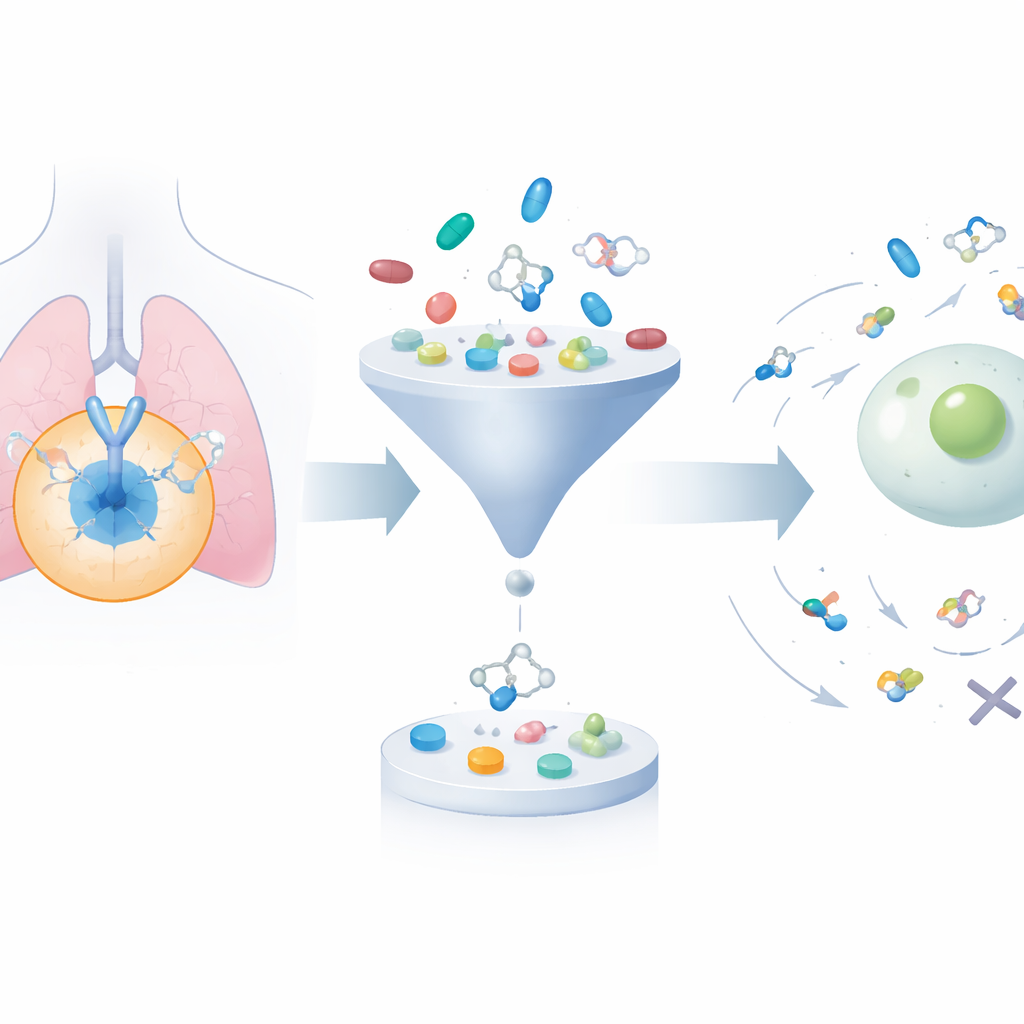
Ampliar y podar una vasta biblioteca de moléculas
El marco comienza recopilando alrededor de 24.000 moléculas conocidas que bloquean EGFR a partir de bases de datos públicas. Luego utiliza un algoritmo generativo para modificar sistemáticamente sus estructuras, produciendo aproximadamente 200.000 candidatas relacionadas. Se aplican dos filtros bien establecidos de «likeliness» farmacéutico para deshacerse de compuestos que son demasiado voluminosos, demasiado grasos o que, por otras razones, probablemente no se comporten bien en el organismo, reduciendo el conjunto a menos de 50.000. A continuación, un programa de acoplamiento molecular (docking) ajusta virtualmente cada molécula en el bolsillo tridimensional de EGFR donde los fármacos reales se unirían, estimando qué tan fuerte podría fijarse cada una. Esto centra la atención en compuestos que son tanto químicamente razonables como predichos para interactuar bien con la diana.
Enseñar a los ordenadores a reconocer señales de advertencia de toxicidad
Unirse a EGFR es solo la mitad de la historia; un compuesto prometedor también debe evitar dañar tejidos sanos. Para estimar la toxicidad, el estudio recurre a un gran conjunto de datos público llamado Tox21, que registra cómo más de 10.000 químicos afectan diversas vías celulares. Los autores se centran en una vía vinculada al receptor de andrógenos, elegida porque está bien anotada y es biológicamente relevante para varios cánceres. Cada molécula de Tox21 se traduce en una huella digital numérica que captura sus características estructurales y similitudes con otros químicos. Estas huellas alimentan varios modelos predictivos, incluidos redes neuronales, árboles de decisión, una máquina de vectores de soporte tradicional y una máquina de vectores de soporte inspirada en lo cuántico que utiliza un circuito cuántico simple para comparar compuestos en un espacio matemático distinto.
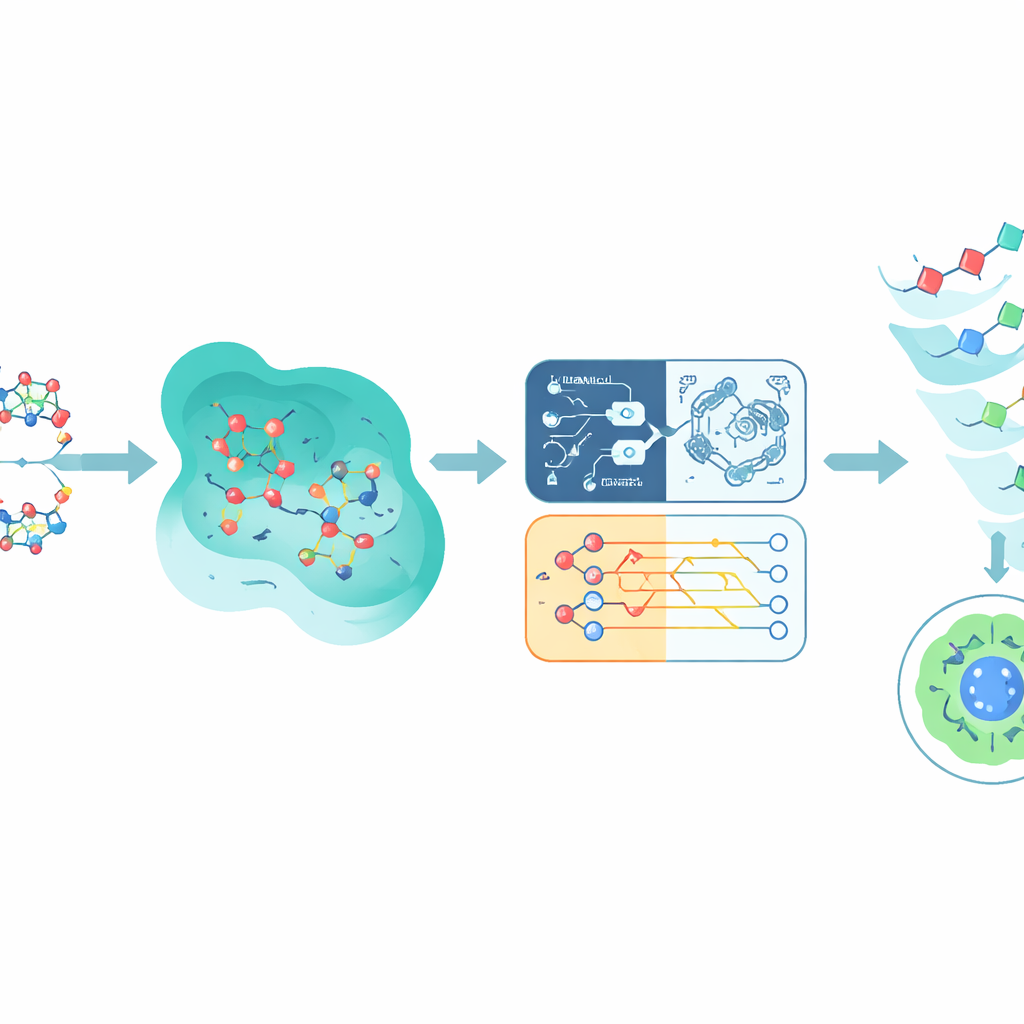
Mezclando predicciones cuánticas y clásicas
En lugar de apostar por un único modelo, Q-CaDD combina las salidas de los cuatro en un conjunto (ensemble), dando mayor peso a la red neuronal pero aun así incorporando la señal más débil pero distinta del modelo cuántico. Cuando se prueba con datos de Tox21 no vistos previamente, este enfoque combinado supera a cualquier modelo individual para distinguir compuestos más y menos tóxicos, medido por una puntuación estándar de clasificación llamada área bajo la curva ROC. Aunque la mejora es modesta y la parte cuántica aún se ejecuta en un simulador en lugar de un chip cuántico real, los resultados sugieren que métodos inspirados en lo cuántico pueden añadir matices útiles a los flujos de trabajo de aprendizaje automático existentes incluso en sus fases iniciales.
De las fichas de puntuación computacionales a las pruebas de laboratorio futuras
Tras validar los modelos de toxicidad, los autores aplican la canalización completa de Q-CaDD a la biblioteca filtrada centrada en EGFR. Evitan emitir un veredicto tajante de sí o no sobre la toxicidad; en su lugar mantienen puntuaciones de riesgo continuas y las combinan con las estimaciones de acoplamiento de la fuerza de unión. Esto produce una lista priorizada de moléculas candidatas, algunas de las cuales parecen unirse a EGFR más fuertemente que un fármaco de referencia mientras conservan una baja toxicidad predicha. Estas moléculas no se presentan como nuevos medicamentos; se señalan como leads que merecen pruebas en el laboratorio. La principal conclusión del estudio para el público general no es que los ordenadores cuánticos ya hayan revolucionado el descubrimiento de fármacos, sino que híbridos cuidadosamente diseñados de herramientas clásicas e inspiradas en lo cuántico pueden ya ayudar a afinar la búsqueda, orientando a los investigadores hacia mejores candidatos con mayor rapidez y manteniendo expectativas realistas sobre los límites del hardware actual.
Cita: Badarala, L. Q-CaDD: accelerating in silico methodologies with quantum computation and machine learning for Epidermal growth factor receptor. Sci Rep 16, 14436 (2026). https://doi.org/10.1038/s41598-026-44978-4
Palabras clave: descubrimiento de fármacos cuántico, inhibidores de EGFR, toxicidad y aprendizaje automático, cribado virtual, cáncer de pulmón no microcítico