Clear Sky Science · sv
Förutsägelse av circRNA:s subcellulära lokalisation genom att sammanföra circRNA-sekvens och nätverksinformation
Varför små RNA-loopar och deras adresser spelar roll
Inuti varje mänsklig cell rör sig ett stort antal RNA-molekyler och hjälper till att styra vilka gener som slås på och av. Bland dem finns cirkulära RNA, eller circRNA—ovanliga loopformade RNA-segment som är förvånansvärt stabila och nära kopplade till många sjukdomar, inklusive cancer. För att förstå vad dessa molekyler gör behöver forskare en grundläggande uppgift: var i cellen de befinner sig. Att kartlägga circRNA:s ”adresser” med traditionella laboratorieexperiment är dock långsamt, kostsamt och ofullständigt. Denna studie presenterar en ny datorbaserad metod, kallad CircLoc, som förutspår var circRNA:n finns i cellen genom att kombinera information från deras sekvenser och från de komplexa biologiska nätverk de ingår i.
Små loopar med stora biologiska roller
Tidigare avfärdade som ofarliga rester från genbearbetning är circRNA nu kända för att påverka en rad viktiga processer, från hur celler differentierar sig till hur gener regleras. Deras cirkulära form gör dem mer stabila än många andra RNA, vilket i sin tur gör dem intressanta som potentiella biomarkörer för sjukdomsdiagnostik. CircRNA kan binda proteiner och absorbera mikroRNA—små regulatorer som normalt dämpar genaktivitet—och därigenom förändra cellulärt beteende. Eftersom många molekyler verkar endast i specifika delar av cellen, såsom kärnan, cytoplasman eller membranen, ger kunskap om ett circRNA:s subcellulära lokalisation viktiga ledtrådar till dess funktion och möjliga roll i hälsa och sjukdom.
Att omvandla spridda data till en träningsmiljö
Författarna började med att sätta samman en noggrant kurerad samling mänskliga circRNA med kända lokalisationer från flera offentliga databaser. Efter att ha tagit bort sällsynta kategorier och extremt obalanserade grupper fokuserade de på sju huvudsakliga cellulära områden, inklusive kärnan, nukleolen, nukleoplasman, cytoplasman, cytosolen, kromatin och membran. Totalt samlade de 1 486 circRNA med pålitlig sekvensinformation och minst en känd lokalisation; många tillhörde flera regioner samtidigt, vilket gjorde uppgiften till ett verkligt flermärkesproblem. Ytterligare dataset från tidigare databasutgåvor och från en stor cancerrelaterad samling reserverades som oberoende tester, vilket gjorde det möjligt för teamet att undersöka hur väl deras modell generaliserar till nyligen rapporterade circRNA.
Att blanda sekvensmönster med kartor över cellulära interaktioner
CircLocs kärnidé är att ett circRNA:s adress formas inte bara av dess egen sekvens utan också av vilket sällskap det håller. På sekvenssidan undersöker modellen korta sekvensfragment (k-mer och deras reverskomplement) och rikare mönster inlärda av en stor RNA-fokuserad språkmodell kallad RNAErnie, som ursprungligen tränades på massiva RNA-dataset för att fånga subtila regelbundenheter. På nätverkssidan byggde författarna flera kartor som visar hur circRNA kopplar till varandra och till relaterade biologiska entiteter: överlappande sekvenser, associerade sjukdomar, läkemedelssvar, interagerande mikroRNA och bindande proteiner. Ett nätverksinbäddningsverktyg kallat node2vec omvandlar strukturen i varje karta till numeriska funktioner, och en grafuppmärksamhetsautoencoder (GATE) förfinar dem sedan genom att betona kopplingar mellan circRNA som beter sig likartat, vilket effektivt reducerar brus och berikar de nätverksbaserade signalerna.
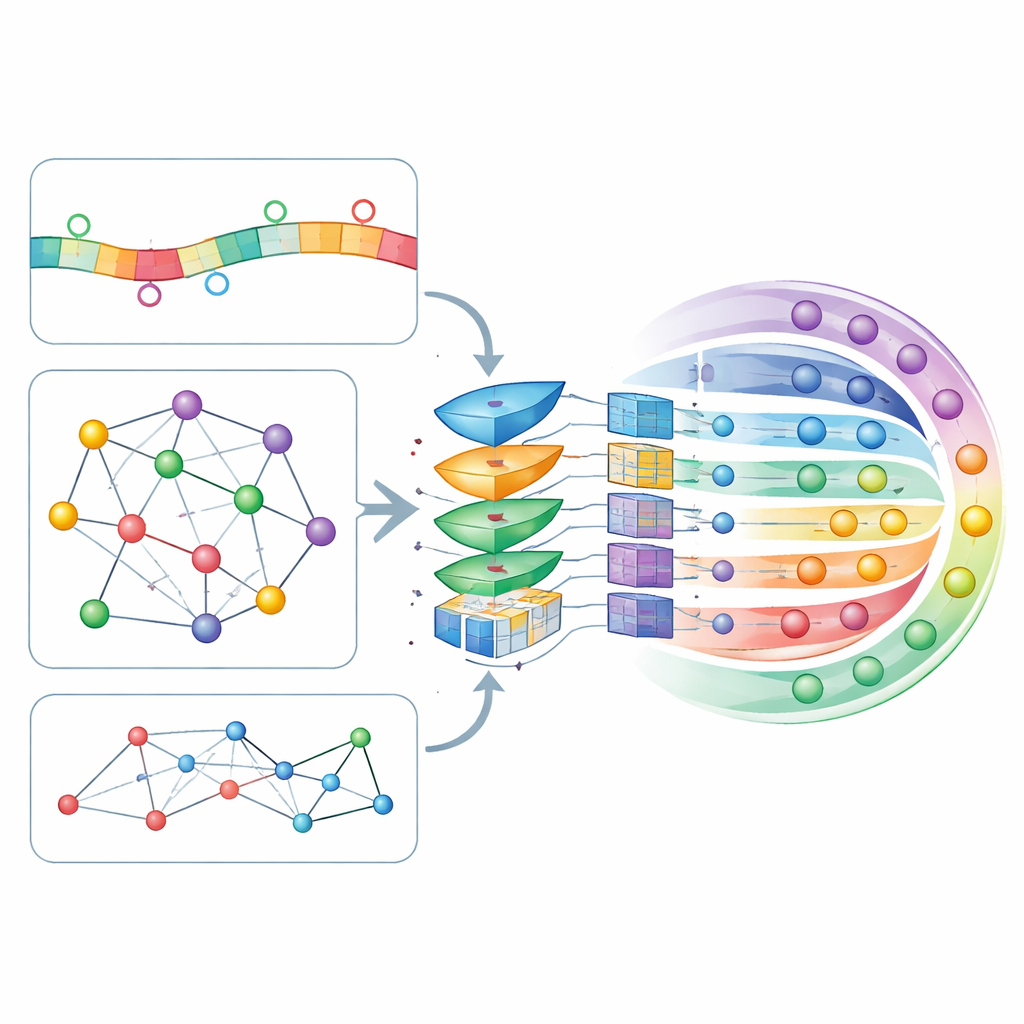
Låta modellen avgöra vad som betyder mest
Alla dessa sekvens- och nätverksbaserade funktioner sys ihop till en enda profil för varje circRNA och skickas genom ett självuppmärksamhetslager, en mekanism som låter modellen lära sig vilka kombinationer av funktioner som bör påverka dess beslut mest. De förfinade profilerna matas sedan in i ett djupt, fullständigt uppkopplat neuralt nätverk som ger en sannolikhet för var och en av de sju möjliga lokalisationerna. Författarna finjusterade modellens många inställningar med tiofaldig korsvalidering, en rigorös procedur som upprepade gånger delar upp data i tränings- och testdelar. CircLoc uppnådde ett genomsnittligt värde på omkring 0,79 på ett standardmått för prestanda (AUC), och överträffade tydligt tidigare metoder utformade för mikroRNA samt klassiska flermärkmetoder tränade på samma funktioner. Experiment som tog bort specifika funktioner eller moduler visade att nätverksinformationen och GATE-förfiningen var särskilt viktiga, medan sekvensfunktionerna fortfarande bidrog med användbara, om än mindre, förbättringar.
Hur väl hanterar modellen nya circRNA?
För att pröva verklig användbarhet tränade teamet CircLoc på en version av lokaliseringsdatabasen och testade den på circRNA som bara förekom i en senare utgåva, samt på en separat cancerfokuserad resurs. Prestandan sjönk jämfört med den ursprungliga träningsuppsättningen, vilket var väntat när man möter genuint nya data från andra källor, men höll sig ändå respektabel: genomsnittspoängen minskade måttligt men visade fortfarande meningsfull prediktiv förmåga. Dessa tester, tillsammans med jämförelser med andra metoder, tyder på att CircLoc kan ge rimliga första uppskattningar för lokalisationen av nyupptäckta circRNA, även när viss stödinformation—såsom detaljerade sjukdoms- eller läkemedelsassociationer—saknas.
Vad detta betyder för framtida RNA-forskning
Detta arbete visar att kombinationen av direkt sekvensinformation och rika interaktionsnätverk kan hjälpa beräkningsmodeller att förutse var circRNA sannolikt finns i en cell. För experimentella biologer erbjuder CircLoc ett sätt att prioritera vilka circRNA som bör studeras i vilka cellulära kompartment, vilket potentiellt sparar tid och resurser. Även om metoden ännu inte kan ersätta laboratoriemätningar, och dess skapare påpekar begränsningar såsom ofullständiga data och måttlig prestanda i vissa testuppsättningar, utgör den ett viktigt steg mot storskaliga, in silico ”adressböcker” för RNA-molekyler. Allteftersom databaser växer och modelleringsmetoder förbättras kan sådana verktyg bli rutinmässiga följeslagare till experiment och vägleda sökandet efter de circRNA som är mest relevanta för sjukdom och terapi.
Citering: Chen, L., Hu, J. & Zhou, B. Predicting circRNA subcellular localization by fusing circRNA sequence and network information. Sci Rep 16, 12775 (2026). https://doi.org/10.1038/s41598-026-43808-x
Nyckelord: cirkulärt RNA, subcellulär lokalisation, beräkningsbiologi, maskininlärning, RNA-nätverk