Clear Sky Science · nl
Voorspellen van de subcellulaire lokalisatie van circRNA door het samenvoegen van circRNA-sequentie- en netwerkinformatie
Waarom kleine RNA-lussen en hun adressen ertoe doen
In elke menselijke cel bewegen talloze RNA-moleculen voortdurend, en ze helpen bepalen welke genen aan- of uitgezet worden. Tot deze groep behoren circulaire RNA’s, of circRNA’s—ongewone lusvormige RNA-fragmenten die opmerkelijk stabiel zijn en sterk verbonden zijn met veel ziekten, waaronder kanker. Om te begrijpen wat deze moleculen doen, moeten onderzoekers een basale vraag beantwoorden: waar in de cel bevinden ze zich? Het in kaart brengen van de "adressen" van circRNA’s met traditionele laboratoriumexperimenten is echter traag, kostbaar en vaak incompleet. Deze studie presenteert een nieuwe computergebaseerde methode, CircLoc, die voorspelt waar circRNA’s in de cel voorkomen door informatie uit hun sequenties te combineren met gegevens uit de complexe biologische netwerken waarin ze meespelen.
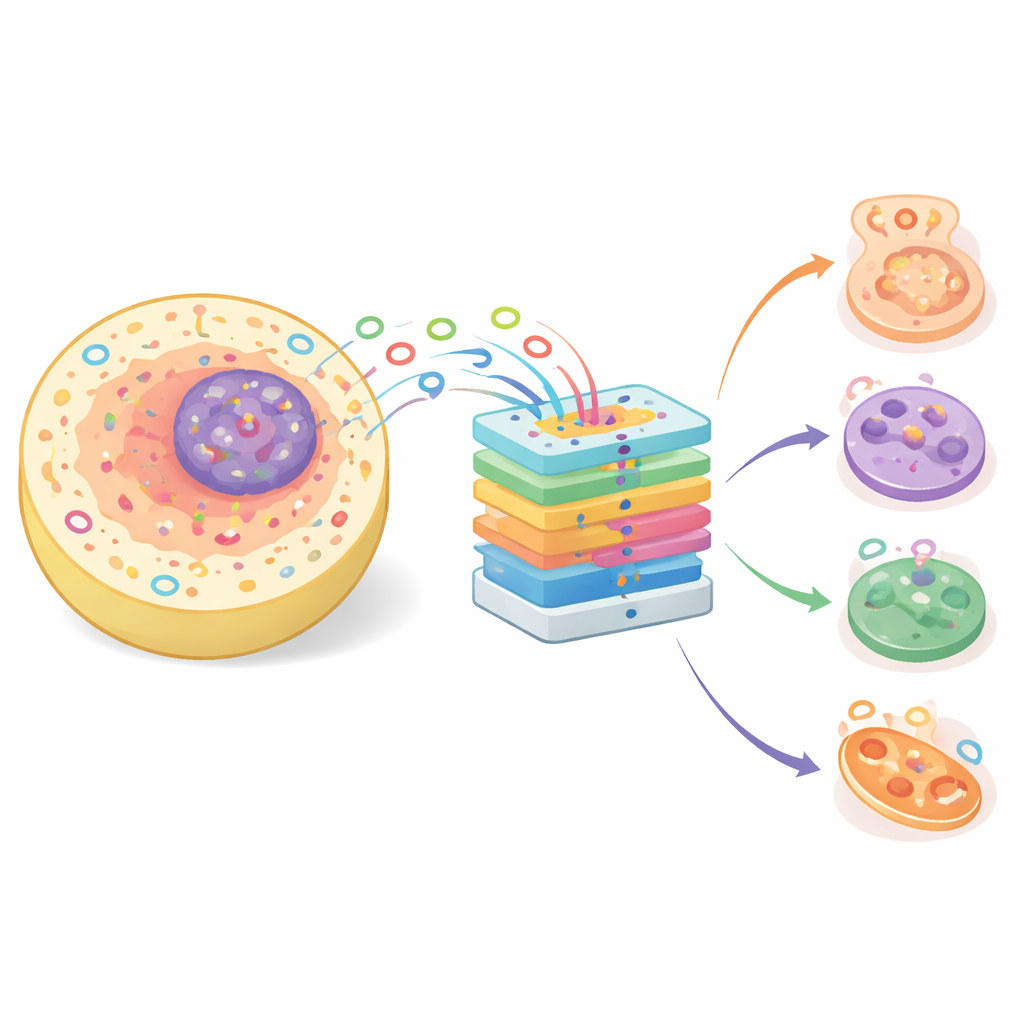
Kleine lussen met grote biologische rollen
Die vroeger werden afgedaan als nutteloze bijproducten van genbewerking, maar circRNA’s blijken nu invloed te hebben op een reeks van cruciale processen, van celdifferentiatie tot genregulatie. Hun circulaire vorm maakt ze stabieler dan veel andere RNA’s, waardoor ze aantrekkelijk worden als potentiële biomarkers voor diagnostiek. CircRNA’s kunnen eiwitten binden en microRNA’s—kleine regulatoren die normaal gezien genactiviteit dempen—opslokken, en zo het cellulaire gedrag herstructureren. Omdat veel moleculen alleen in specifieke compartimenten van de cel actief zijn, zoals de kern, het cytoplasma of membranen, biedt kennis over de subcellulaire locatie van een circRNA belangrijke aanwijzingen voor zijn functie en mogelijke rol bij gezondheid en ziekte.
Versnipperde gegevens omzetten in een trainingsbasis
De auteurs begonnen met het samenstellen van een zorgvuldig gecureerde verzameling humane circRNA’s met bekende lokalisaties uit verschillende openbare databases. Na het verwijderen van zeldzame categorieën en extreem onevenwichtige groepen concentreerden ze zich op zeven hoofdcelregio’s, waaronder kern, nucleolus, nucleoplasma, cytoplasma, cytosol, chromatine en membranen. In totaal verzamelden ze 1.486 circRNA’s met betrouwbare sequentie-informatie en ten minste één bekende locatie; veel exemplaren behoorden tegelijk tot meerdere regio’s, waardoor het een echte multi-label voorspellingsopgave werd. Extra datasets uit eerdere database-releases en uit een grote kankergerelateerde collectie werden opzij gezet als onafhankelijke tests, zodat het team kon onderzoeken hoe goed hun model zou generaliseren naar nieuw gerapporteerde circRNA’s.
Sequentiepatronen mengen met cellulaire interactiekaarten
Het kernidee van CircLoc is dat het adres van een circRNA niet alleen door zijn eigen sequentie wordt bepaald, maar ook door de ‘maatschappij’ waarin het verkeert. Aan de sequentiekant bekijkt het model korte sequentiefragmenten (k-mers en hun reverse complements) en rijkere patronen die zijn geleerd door een groot RNA-georiënteerd taalmodel genaamd RNAErnie, dat oorspronkelijk op enorme RNA-datasets is getraind om subtiele regelmatigheden vast te leggen. Aan de netwerkzijde bouwden de auteurs meerdere kaarten die laten zien hoe circRNA’s met elkaar en met gerelateerde biologische entiteiten verbonden zijn: overlappende sequenties, geassocieerde ziekten, geneesmiddelreacties, interagerende microRNA’s en bindende eiwitten. Een netwerk-embeddingtool genaamd node2vec zet de structuur van elke kaart om in numerieke kenmerken, en een graph attention auto-encoder (GATE) verfijnt die kenmerken vervolgens door verbindingen tussen circRNA’s die zich vergelijkbaar gedragen te benadrukken, waarmee de netwerkafgeleide signalen effectief worden ontdaan van ruis en verrijkt.
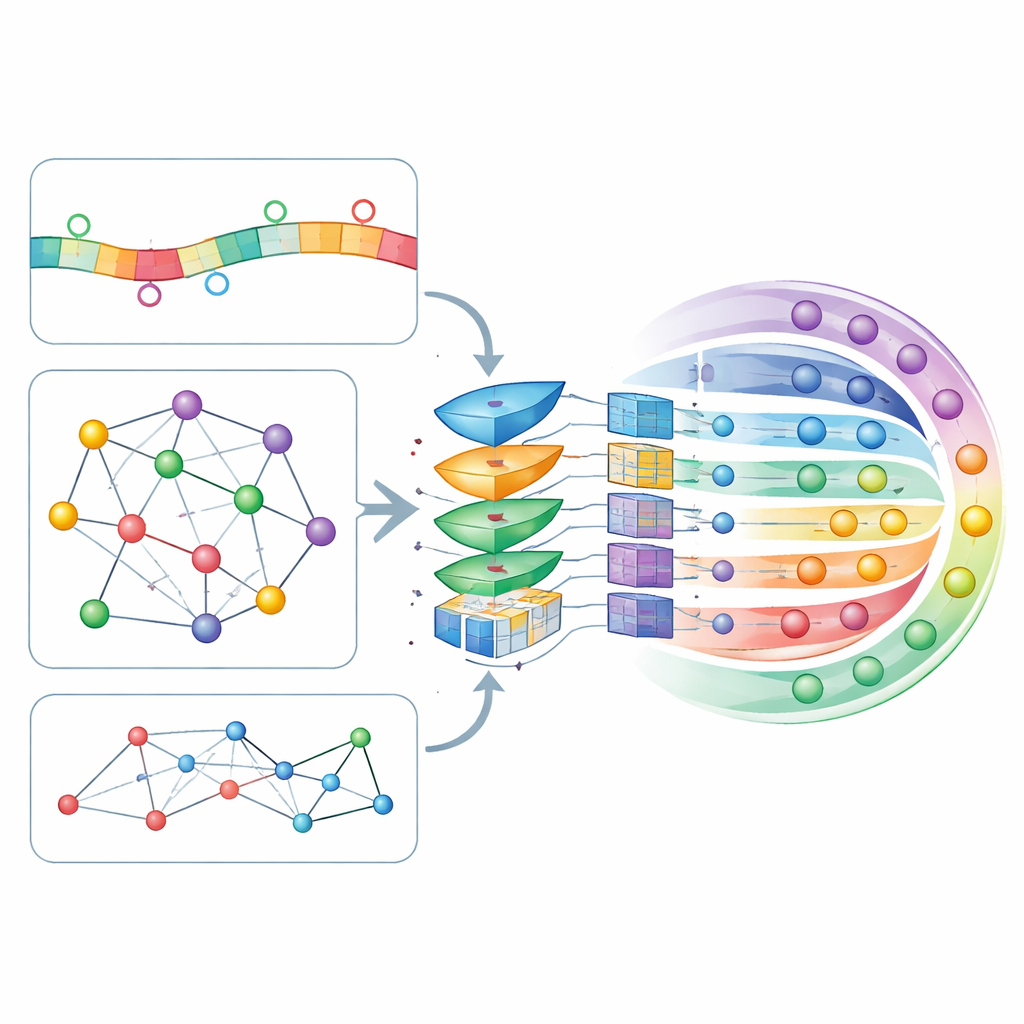
Het model laten beslissen wat het belangrijkst is
Al deze sequentie- en netwerkgebaseerde kenmerken worden samengevoegd tot een enkel profiel voor elk circRNA en door een self-attentionlaag geleid, een mechanisme waarmee het model kan leren welke combinaties van kenmerken het sterkst moeten meewegen in zijn beslissingen. De verfijnde profielen worden daarna ingevoerd in een diepe, volledig verbonden neurale netwerk dat voor elk van de zeven mogelijke locaties een waarschijnlijkheid uitspoort. De auteurs stemden de vele instellingen van het model af met tienvoudige cross-validatie, een rigoureuze procedure die de data herhaaldelijk in trainings- en testdelen verdeelt. CircLoc behaalde een gemiddelde score van ongeveer 0,79 op een standaard kwaliteitsmaat (AUC) en overtrof daarmee duidelijk eerdere benaderingen die waren ontworpen voor microRNA’s en klassieke multi-label methoden die op dezelfde kenmerken waren getraind. Experimenten waarin specifieke kenmerken of modules werden verwijderd toonden aan dat de netwerkinformatie en de GATE-verfijning bijzonder belangrijk waren, terwijl sequentiekenmerken nog steeds nuttige, zij het kleinere, verbeteringen bijdroegen.
Hoe gaat het model om met nieuwe circRNA’s?
Om bruikbaarheid in de praktijk te testen, trainde het team CircLoc op één versie van de lokalisatiedatabase en testte het model op circRNA’s die alleen in een latere release verschenen, evenals op een afzonderlijke, kankergerichte bron. De prestaties daalden vergeleken met de oorspronkelijke trainingsset, wat te verwachten is bij werkelijk nieuwe data uit andere bronnen, maar bleven respectabel: de gemiddelde scores namen bescheiden af maar gaven nog steeds betekenisvolle voorspellende kracht aan. Deze tests, samen met vergelijkingen met andere methoden, suggereren dat CircLoc redelijke eerste inschattingen kan geven voor de locaties van nieuw ontdekte circRNA’s, zelfs wanneer sommige ondersteunende informatie—zoals gedetailleerde ziekte- of geneesmiddelassociaties—ontbreekt.
Wat dit betekent voor toekomstig RNA-onderzoek
Dit werk laat zien dat het combineren van directe sequentie-informatie met rijke interactienetwerken computationele modellen kan helpen voorspellen waar circRNA’s zich waarschijnlijk in een cel bevinden. Voor experimentele biologen biedt CircLoc een manier om te prioriteren welke circRNA’s in welke cellulaire compartimenten verder onderzocht moeten worden, wat mogelijk tijd en middelen bespaart. Hoewel de methode nog geen laboratoriummetingen kan vervangen, en de makers beperkingen noemen zoals onvolledige data en bescheiden prestaties op sommige testsets, vormt het een belangrijke stap richting grootschalige, in silico "adressenboeken" voor RNA-moleculen. Naarmate databases groeien en modelleertechnieken verbeteren, kunnen dergelijke hulpmiddelen routinepartners bij experimenten worden en het zoeken naar de circRNA’s die het meest relevant zijn voor ziekte en therapie sturen.
Bronvermelding: Chen, L., Hu, J. & Zhou, B. Predicting circRNA subcellular localization by fusing circRNA sequence and network information. Sci Rep 16, 12775 (2026). https://doi.org/10.1038/s41598-026-43808-x
Trefwoorden: circulair RNA, subcellulaire lokalisatie, computationele biologie, machine learning, RNA-netwerken