Clear Sky Science · it
Predire la localizzazione subcellulare delle circRNA fondendo sequenza e informazioni di rete
Perché piccoli anelli di RNA e i loro indirizzi contano
All’interno di ogni cellula umana, vasti numeri di molecole di RNA sono in continuo movimento, contribuendo a controllare quali geni si attivano o si spengono. Tra queste ci sono le RNA circolari, o circRNA—porzioni di RNA dalla forma ad anello, sorprendentemente stabili e strettamente legate a numerose malattie, compresi i tumori. Per capire cosa facciano queste molecole, gli scienziati devono conoscere un fatto fondamentale: dove si trovano nella cellula. Tuttavia mappare gli “indirizzi” delle circRNA con esperimenti di laboratorio tradizionali è lento, costoso e incompleto. Questo studio presenta un nuovo metodo informatico, chiamato CircLoc, che predice dove risiedono le circRNA all’interno delle cellule combinando informazioni dalle loro sequenze e dalle complesse reti biologiche di cui fanno parte.
Piccoli anelli con grandi ruoli biologici
Un tempo considerate scarti innocui del processamento genico, oggi si sa che le circRNA influenzano una gamma di processi vitali, dalla differenziazione cellulare alla regolazione genica. La loro forma circolare le rende più stabili rispetto a molte altre RNA, il che le rende promettenti come potenziali biomarcatori per la diagnosi di malattie. Le circRNA possono legare proteine e sequestrare microRNA—piccoli regolatori che normalmente attenuano l’attività genica—rimodellando così il comportamento cellulare. Poiché molte molecole agiscono solo in parti specifiche della cellula, come il nucleo, il citoplasma o le membrane, conoscere la localizzazione subcellulare di una circRNA fornisce indizi importanti sulla sua funzione e sul suo possibile ruolo in salute e malattia.
Trasformare dati dispersi in un terreno di addestramento
Gli autori hanno iniziato assemblando una collezione accuratamente curata di circRNA umane con localizzazioni note provenienti da diversi database pubblici. Dopo aver rimosso categorie rare e gruppi fortemente sbilanciati, si sono concentrati su sette regioni cellulari principali, tra cui nucleo, nucleolo, nucleoplasma, citoplasma, citosol, cromatina e membrane. In totale hanno raccolto 1.486 circRNA con informazioni di sequenza affidabili e almeno una localizzazione nota; molte appartenevano contemporaneamente a più regioni, rendendo il compito un vero problema di previsione multilabel. Ulteriori set di dati provenienti da rilasci precedenti dei database e da una grande raccolta correlata al cancro sono stati messi da parte come test indipendenti, permettendo al gruppo di valutare quanto bene il modello potesse generalizzare su circRNA segnalate successivamente.
Unire schemi di sequenza con mappe di interazione cellulare
L’idea centrale di CircLoc è che l’indirizzo di una circRNA sia determinato non solo dalla sua sequenza, ma anche dall’ambiente con cui interagisce. Sul versante della sequenza, il modello esamina brevi frammenti (k-mer e i loro complementi inversi) e schemi più ricchi appresi da un grande modello linguistico focalizzato sugli RNA chiamato RNAErnie, originariamente addestrato su vasti dataset di RNA per catturare regolarità sottili. Sul versante della rete, gli autori hanno costruito varie mappe che mostrano come le circRNA si collegano tra loro e ad entità biologiche correlate: sovrapposizione di sequenze, malattie associate, risposte a farmaci, microRNA interagenti e proteine leganti. Uno strumento di embedding di rete chiamato node2vec converte la struttura di ogni mappa in caratteristiche numeriche, e un auto-encoder grafico con attenzione (GATE) le affina poi enfatizzando le connessioni tra circRNA che si comportano in modo simile, denoising e arricchendo effettivamente i segnali derivati dalla rete.
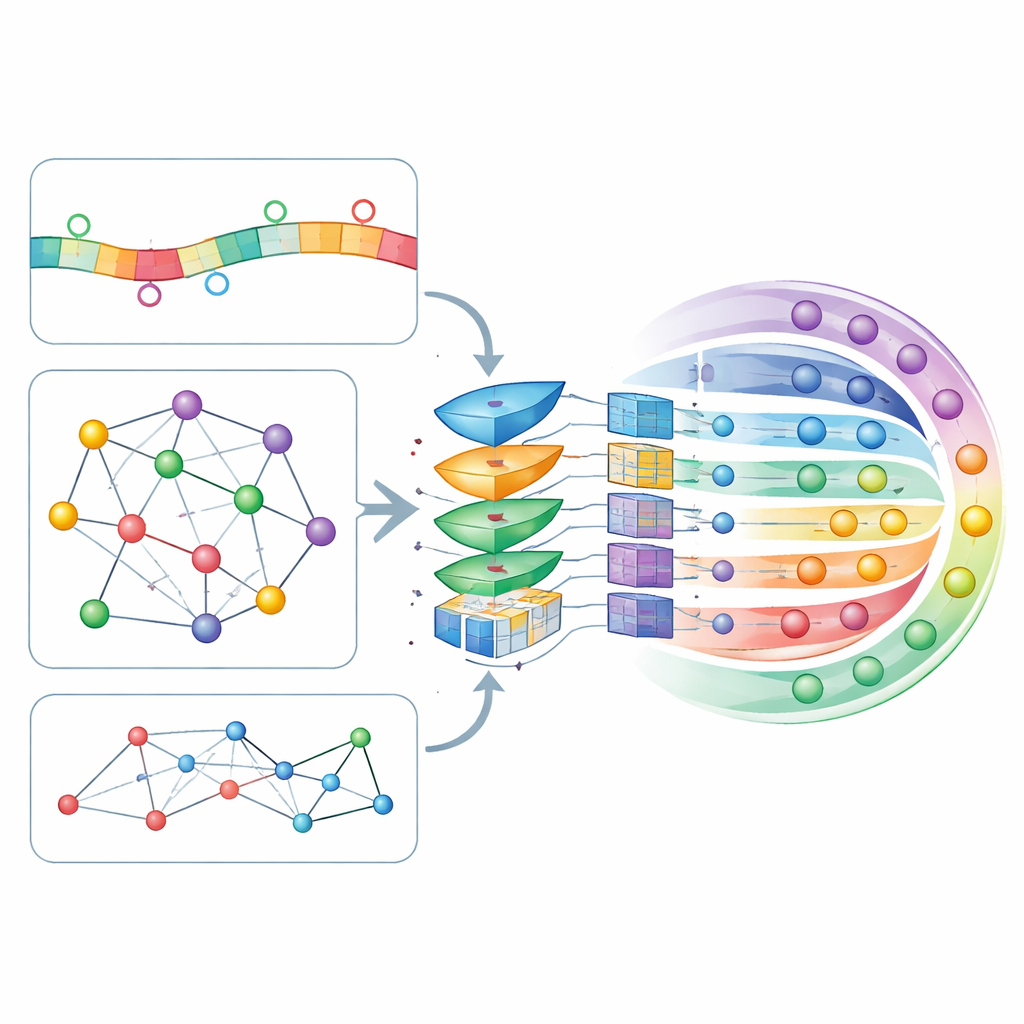
Lasciare che il modello decida cosa conta di più
Tutte queste caratteristiche basate su sequenza e rete vengono assemblate in un unico profilo per ogni circRNA e passate attraverso uno strato di self-attention, un meccanismo che permette al modello di apprendere quali combinazioni di caratteristiche dovrebbero influenzare maggiormente le sue decisioni. I profili raffinati entrano quindi in una rete neurale profonda, completamente connessa, che restituisce una probabilità per ciascuna delle sette possibili localizzazioni. Gli autori hanno ottimizzato i numerosi parametri del modello usando una validazione incrociata a dieci fold, una procedura rigorosa che suddivide ripetutamente i dati in porzioni di addestramento e test. CircLoc ha ottenuto un punteggio medio di circa 0,79 su una misura standard di qualità (AUC), superando nettamente gli approcci precedenti progettati per i microRNA e i metodi multilabel classici addestrati sulle stesse caratteristiche. Esperimenti che hanno rimosso specifiche feature o moduli hanno mostrato che le informazioni di rete e la raffinazione tramite GATE erano particolarmente importanti, mentre le caratteristiche di sequenza fornivano comunque miglioramenti utili, sebbene più modesti.
Quanto bene il modello gestisce nuove circRNA?
Per sondare l’utilità nel mondo reale, il team ha addestrato CircLoc su una versione del database di localizzazione e lo ha testato su circRNA apparse solo in un rilascio successivo, oltre che su una risorsa separata focalizzata sul cancro. Le prestazioni sono calate rispetto al set di addestramento originale, come ci si aspetta di fronte a dati realmente nuovi provenienti da fonti diverse, ma sono rimaste rispettabili: i punteggi medi sono diminuiti modestamente pur indicando una potenza predittiva significativa. Questi test, insieme ai confronti con altri metodi, suggeriscono che CircLoc può fornire ipotesi ragionevoli di primo livello sulle localizzazioni di circRNA recentemente scoperte, anche quando alcune informazioni di supporto—come dettagliate associazioni con malattie o farmaci—mancano.
Cosa significa per la ricerca futura sugli RNA
Questo lavoro dimostra che combinare informazioni dirette di sequenza con ricche reti di interazione può aiutare i modelli computazionali a prevedere dove le circRNA sono più probabilmente localizzate all’interno di una cellula. Per i biologi sperimentali, CircLoc offre un modo per dare priorità a quali circRNA studiare in quali compartimenti cellulari, potenzialmente risparmiando tempo e risorse. Sebbene il metodo non possa ancora sostituire le misurazioni di laboratorio, e i suoi creatori segnalino limiti come dati incompleti e prestazioni modeste su alcuni set di test, rappresenta un passo importante verso grossi “elenco indirizzi” in silico per le molecole di RNA. Con la crescita dei database e il miglioramento delle tecniche di modellazione, tali strumenti potrebbero diventare compagni di routine per gli esperimenti, guidando la ricerca delle circRNA più rilevanti in malattia e terapia.
Citazione: Chen, L., Hu, J. & Zhou, B. Predicting circRNA subcellular localization by fusing circRNA sequence and network information. Sci Rep 16, 12775 (2026). https://doi.org/10.1038/s41598-026-43808-x
Parole chiave: RNA circolare, localizzazione subcellulare, biologia computazionale, apprendimento automatico, reti di RNA