Clear Sky Science · pt
Predizendo a localização subcelular de circRNA ao fundir sequência de circRNA e informação de rede
Por que pequenos laços de RNA e seus endereços importam
Dentro de cada célula humana, vastas quantidades de moléculas de RNA circulam, ajudando a controlar quais genes são ativados ou silenciados. Entre elas estão os RNAs circulares, ou circRNAs — fragmentos de RNA em forma de laço que são surpreendentemente estáveis e estão fortemente ligados a muitas doenças, incluindo cânceres. Para entender o que essas moléculas fazem, os cientistas precisam saber um fato básico: onde na célula elas se localizam. No entanto, mapear os “endereços” dos circRNAs com experimentos de laboratório tradicionais é lento, caro e incompleto. Este estudo apresenta um novo método computacional, chamado CircLoc, que prevê onde os circRNAs residem dentro das células ao combinar informações de suas sequências e das complexas redes biológicas em que participam.
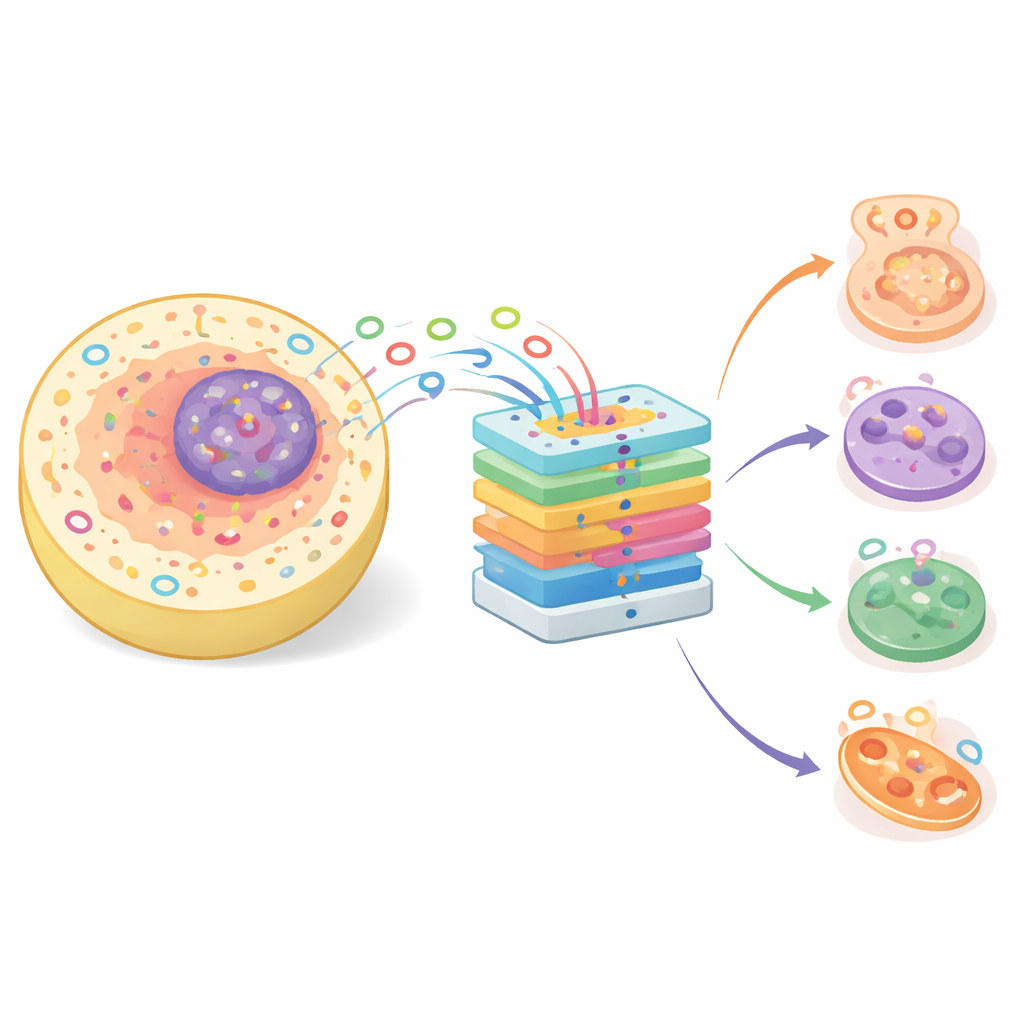
Pequenos laços com grandes papéis biológicos
Antes descartados como sobras inofensivas do processamento gênico, os circRNAs agora são reconhecidos por influenciar uma série de processos vitais, desde a diferenciação celular até a regulação gênica. Sua forma circular os torna mais estáveis do que muitos outros RNAs, o que os torna promissores como potenciais biomarcadores para diagnóstico de doenças. Os circRNAs podem se ligar a proteínas e capturar microRNAs — pequenos reguladores que normalmente atenuam a atividade gênica — remodelando assim o comportamento celular. Como muitas moléculas atuam apenas em partes específicas da célula, como o núcleo, o citoplasma ou membranas, conhecer a localização subcelular de um circRNA fornece pistas importantes sobre sua função e seu possível papel na saúde e na doença.
Transformando dados dispersos em um campo de treinamento
Os autores começaram reunindo uma coleção cuidadosamente curada de circRNAs humanos com locais conhecidos a partir de vários bancos de dados públicos. Após remover categorias raras e grupos extremamente desbalanceados, eles se concentraram em sete regiões celulares principais, incluindo núcleo, nucleolo, nucleoplasma, citoplasma, citosol, cromatina e membranas. No total, reuniram 1.486 circRNAs com informações de sequência confiáveis e pelo menos um local conhecido; muitos pertenciam a múltiplas regiões ao mesmo tempo, tornando a tarefa um verdadeiro problema de predição multilabel. Conjuntos de dados adicionais de versões anteriores dos bancos de dados e de uma grande coleção relacionada ao câncer foram reservados como testes independentes, permitindo à equipe examinar quão bem seu modelo generalizaria para circRNAs relatados posteriormente.
Mesclando padrões de sequência com mapas de interação celular
A ideia central do CircLoc é que o “endereço” de um circRNA é definido não apenas por sua própria sequência, mas também pela vizinhança com a qual interage. No lado da sequência, o modelo analisa fragmentos curtos (k-mers e seus complementos reversos) e padrões mais complexos capturados por um grande modelo de linguagem focado em RNA chamado RNAErnie, originalmente treinado em conjuntos massivos de dados de RNA para apreender regularidades sutis. No lado da rede, os autores construíram vários mapas mostrando como circRNAs se conectam entre si e a entidades biológicas relacionadas: sobreposição de sequências, doenças associadas, respostas a fármacos, microRNAs interagentes e proteínas de ligação. Uma ferramenta de embedding de redes chamada node2vec converte a estrutura de cada mapa em características numéricas, e um autoencoder de atenção em grafos (GATE) então as refina enfatizando conexões entre circRNAs que se comportam de maneira semelhante, efetivamente reduzindo ruído e enriquecendo os sinais derivados da rede.
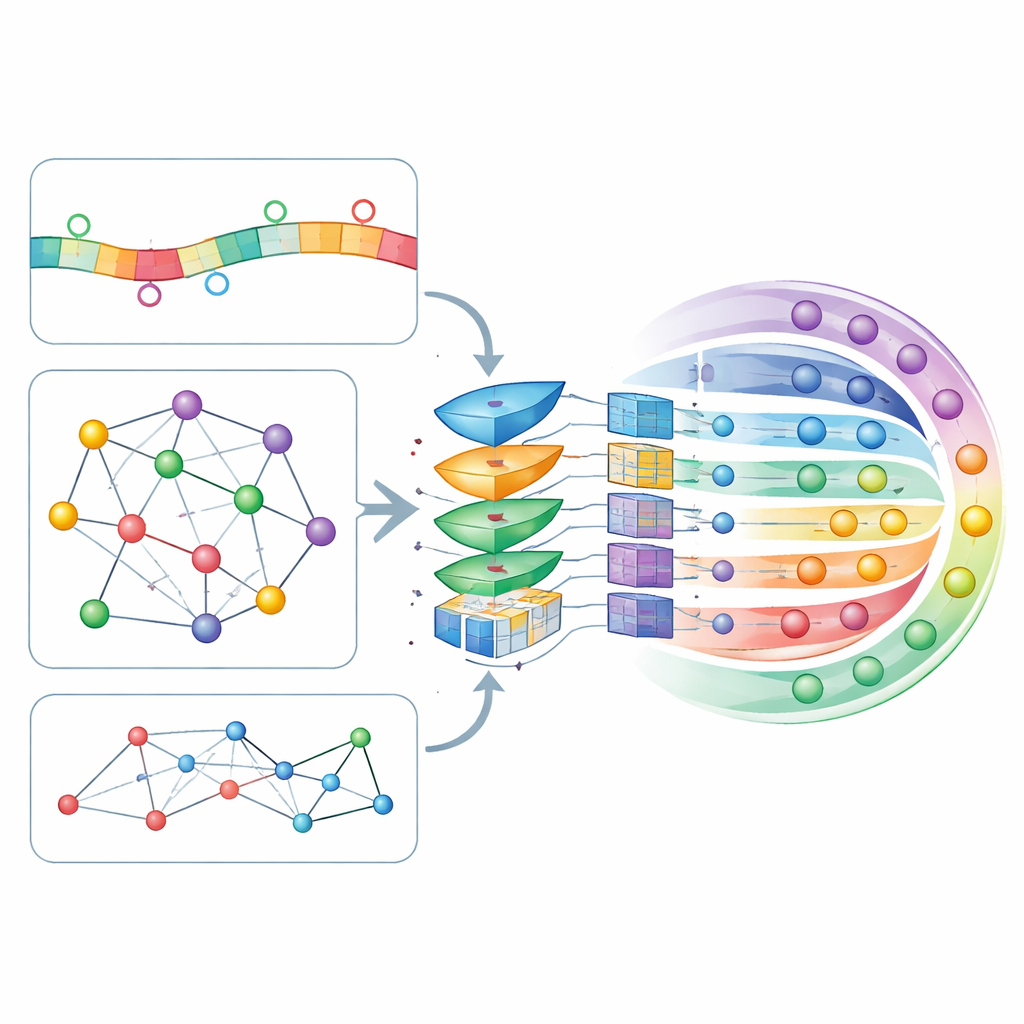
Deixando o modelo decidir o que mais importa
Todas essas características baseadas em sequência e rede são costuradas em um único perfil para cada circRNA e passam por uma camada de auto-atenção, um mecanismo que permite ao modelo aprender quais combinações de características devem influenciar suas decisões com mais força. Os perfis refinados então entram em uma rede neural profunda totalmente conectada que produz uma probabilidade para cada uma das sete localizações possíveis. Os autores ajustaram os muitos parâmetros do modelo usando validação cruzada em dez blocos, um procedimento rigoroso que divide repetidamente os dados em partes de treinamento e teste. O CircLoc alcançou uma pontuação média de cerca de 0,79 em uma medida padrão de qualidade (AUC), superando claramente abordagens anteriores projetadas para microRNAs e métodos multilabel clássicos treinados com as mesmas características. Experimentos que removeram características ou módulos específicos demonstraram que a informação de rede e o refinamento pelo GATE foram particularmente importantes, enquanto as características de sequência ainda contribuíram com melhorias úteis, embora menores.
Quão bem o modelo lida com novos circRNAs?
Para sondar a utilidade no mundo real, a equipe treinou o CircLoc em uma versão do banco de dados de localização e testou em circRNAs que apareceram apenas em uma versão posterior, assim como em um recurso separado focado em câncer. O desempenho caiu em comparação com o conjunto de treinamento original, como esperado ao enfrentar dados genuinamente novos de diferentes fontes, mas permaneceu respeitável: as pontuações médias diminuíram de forma modesta enquanto ainda indicavam poder preditivo significativo. Esses testes, junto com comparações a outros métodos, sugerem que o CircLoc pode fornecer palpites razoáveis de primeira linha sobre as localizações de circRNAs recém-descobertos, mesmo quando algumas informações de suporte — como associações detalhadas com doenças ou fármacos — estão ausentes.
O que isso significa para pesquisas futuras em RNA
Este trabalho mostra que combinar informação direta de sequência com redes de interação ricas pode ajudar modelos computacionais a antecipar onde os circRNAs provavelmente residirão dentro de uma célula. Para biólogos experimentais, o CircLoc oferece uma forma de priorizar quais circRNAs estudar em quais compartimentos celulares, potencialmente economizando tempo e recursos. Embora o método ainda não possa substituir medições de laboratório, e seus criadores apontem limitações como dados incompletos e desempenho modesto em alguns conjuntos de teste, ele representa um passo importante rumo a “listas de endereços” in silico em larga escala para moléculas de RNA. À medida que os bancos de dados crescem e as técnicas de modelagem melhoram, tais ferramentas podem se tornar companheiras rotineiras de experimentos, guiando a busca por circRNAs mais relevantes em doenças e terapias.
Citação: Chen, L., Hu, J. & Zhou, B. Predicting circRNA subcellular localization by fusing circRNA sequence and network information. Sci Rep 16, 12775 (2026). https://doi.org/10.1038/s41598-026-43808-x
Palavras-chave: RNA circular, localização subcelular, biologia computacional, aprendizado de máquina, redes de RNA