Clear Sky Science · es
Predicción de la localización subcelular de circRNA fusionando la secuencia de circRNA y la información de redes
Por qué importan los diminutos bucles de ARN y sus direcciones
Dentro de cada célula humana, un gran número de moléculas de ARN se mueven y colaboran para controlar qué genes se activan o se silencian. Entre ellas están los ARN circulares, o circRNAs: fragmentos de ARN con forma de bucle que son sorprendentemente estables y están estrechamente relacionados con muchas enfermedades, incluidos diversos cánceres. Para entender qué hacen estas moléculas, los científicos necesitan conocer un dato básico: en qué parte de la célula se encuentran. Sin embargo, cartografiar las “direcciones” de los circRNAs con experimentos de laboratorio tradicionales es lento, caro e incompleto. Este estudio presenta un nuevo método informático, llamado CircLoc, que predice dónde residen los circRNAs dentro de la célula combinando información de sus secuencias y de las complejas redes biológicas en las que participan.
Pequeños bucles con grandes roles biológicos
Durante mucho tiempo descartados como restos inofensivos del procesamiento génico, hoy se sabe que los circRNAs influyen en una variedad de procesos vitales, desde la diferenciación celular hasta la regulación génica. Su forma circular los hace más estables que muchos otros ARN, lo que a su vez los convierte en atractivos candidatos como biomarcadores para el diagnóstico de enfermedades. Los circRNAs pueden unirse a proteínas y secuestrar microARNs —pequeños reguladores que normalmente atenúan la actividad génica—, remodelando así el comportamiento celular. Dado que muchas moléculas actúan solo en partes específicas de la célula, como el núcleo, el citoplasma o las membranas, conocer la localización subcelular de un circRNA ofrece pistas importantes sobre su función y su posible papel en la salud y la enfermedad.
Convertir datos dispersos en un terreno de entrenamiento
Los autores empezaron reuniendo una colección cuidadosamente curada de circRNAs humanos con localizaciones conocidas procedentes de varias bases de datos públicas. Tras eliminar categorías raras y grupos extremadamente desequilibrados, se centraron en siete regiones celulares principales, incluida la membrana, el núcleo, el nucleolo, el nucleoplasma, el citoplasma, el citosol y la cromatina. En total reunieron 1.486 circRNAs con información de secuencia fiable y al menos una localización conocida; muchos pertenecían a varias regiones a la vez, lo que convirtió la tarea en un verdadero problema de predicción multilabel. Conjuntos de datos adicionales de versiones anteriores de las bases de datos y una amplia colección relacionada con cáncer se reservaron como pruebas independientes, lo que permitió al equipo evaluar qué tan bien generalizaría su modelo a circRNAs reportados posteriormente.
Mezclar patrones de secuencia con mapas de interacción celular
La idea central de CircLoc es que la «dirección» de un circRNA está determinada no solo por su propia secuencia, sino también por la compañía que mantiene. En el lado de la secuencia, el modelo examina fragmentos cortos (k-mers y sus complementos reversos) y patrones más ricos aprendidos por un gran modelo de lenguaje especializado en ARN llamado RNAErnie, que fue entrenado originalmente con enormes conjuntos de datos de ARN para captar regularidades sutiles. En el lado de la red, los autores construyeron varios mapas que muestran cómo se conectan los circRNAs entre sí y con entidades biológicas relacionadas: solapamiento de secuencias, enfermedades asociadas, respuestas a fármacos, microARNs que interactúan y proteínas de unión. Una herramienta de incrustación de redes llamada node2vec convierte la estructura de cada mapa en características numéricas, y un autoencoder de atención en grafos (GATE) las refina enfatizando las conexiones entre circRNAs que se comportan de forma similar, desruidosando y enriqueciendo eficazmente las señales derivadas de la red.
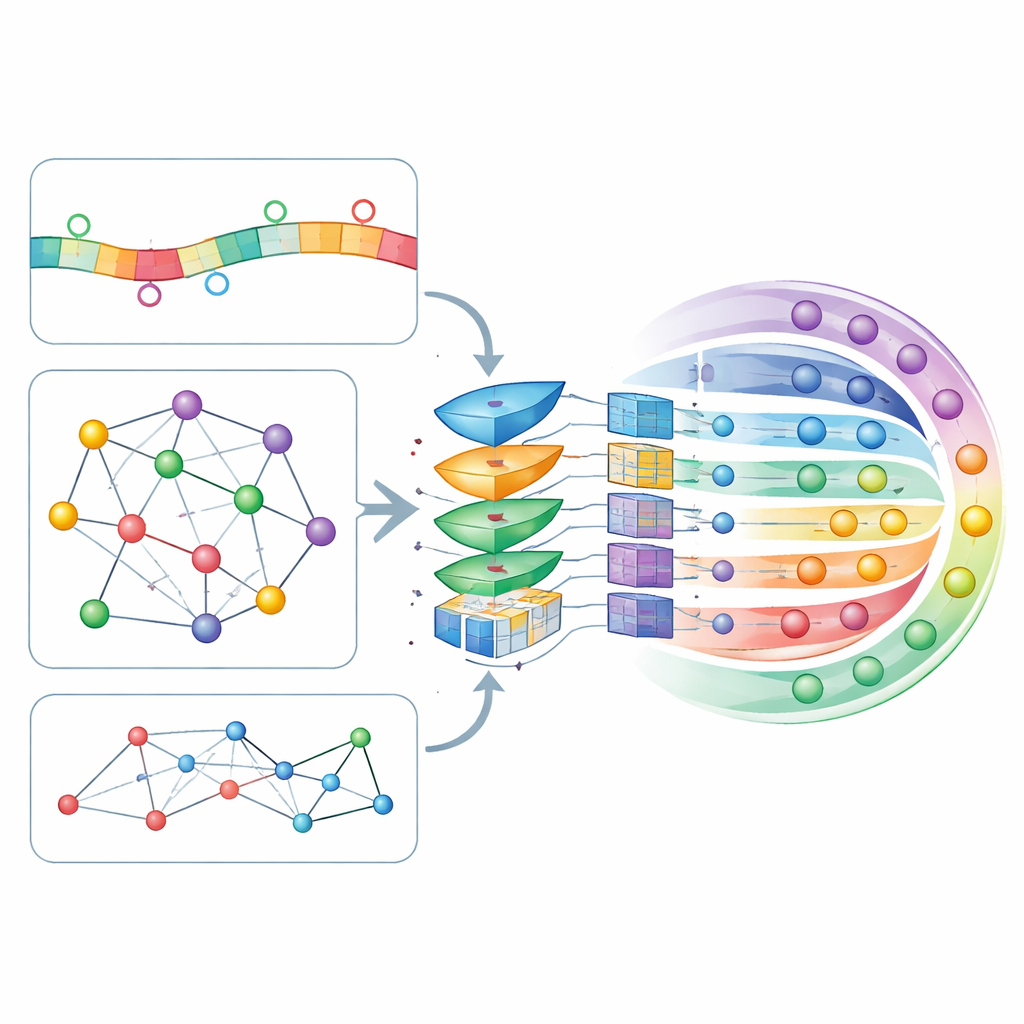
Poner al modelo a decidir qué importa más
Todas estas características basadas en secuencia y red se ensamblan en un perfil único para cada circRNA y se pasan por una capa de autoatención, un mecanismo que permite al modelo aprender qué combinaciones de características deben influir con mayor fuerza en sus decisiones. Los perfiles refinados entran luego en una red neuronal profunda totalmente conectada que produce una probabilidad para cada una de las siete localizaciones posibles. Los autores ajustaron los numerosos parámetros del modelo usando validación cruzada de diez particiones, un procedimiento riguroso que divide repetidamente los datos en porciones de entrenamiento y prueba. CircLoc alcanzó una puntuación media de alrededor de 0,79 en una medida estándar de calidad (AUC), superando con claridad enfoques previos diseñados para microARNs y métodos multilabel clásicos entrenados con las mismas características. Experimentos que eliminaron características o módulos específicos mostraron que la información de la red y la refinación por GATE eran especialmente importantes, mientras que las características de secuencia seguían aportando mejoras útiles, aunque menores.
¿Cómo se comporta el modelo con circRNAs nuevos?
Para evaluar la utilidad en el mundo real, el equipo entrenó CircLoc con una versión de la base de datos de localización y lo probó con circRNAs que aparecieron solo en una versión posterior, así como en un recurso separado centrado en cáncer. El rendimiento bajó en comparación con el conjunto de entrenamiento original, como era de esperar al enfrentarse a datos genuinamente nuevos de fuentes distintas, pero se mantuvo razonable: las puntuaciones medias se redujeron de forma modesta y aún indicaron un poder predictivo significativo. Estas pruebas, junto con comparaciones con otros métodos, sugieren que CircLoc puede ofrecer conjeturas iniciales razonables sobre las localizaciones de circRNAs recién descubiertos, incluso cuando falta parte de la información de apoyo —como asociaciones detalladas con enfermedades o fármacos—.
Qué supone esto para la investigación futura sobre ARN
Este trabajo demuestra que combinar información directa de la secuencia con redes de interacción ricas puede ayudar a los modelos computacionales a anticipar dónde es probable que residan los circRNAs dentro de una célula. Para los biólogos experimentales, CircLoc ofrece una forma de priorizar qué circRNAs estudiar en qué compartimentos celulares, ahorrando potencialmente tiempo y recursos. Aunque el método aún no puede reemplazar las mediciones de laboratorio y sus creadores señalan limitaciones como datos incompletos y rendimiento modesto en algunos conjuntos de prueba, representa un paso importante hacia «libretas de direcciones» in silico a gran escala para moléculas de ARN. A medida que las bases de datos crezcan y las técnicas de modelado mejoren, herramientas de este tipo podrían convertirse en acompañantes habituales de los experimentos, orientando la búsqueda de los circRNAs más relevantes en enfermedad y terapia.
Cita: Chen, L., Hu, J. & Zhou, B. Predicting circRNA subcellular localization by fusing circRNA sequence and network information. Sci Rep 16, 12775 (2026). https://doi.org/10.1038/s41598-026-43808-x
Palabras clave: ARN circular, localización subcelular, biología computacional, aprendizaje automático, redes de ARN