Clear Sky Science · de
Vorhersage der subzellulären Lokalisation von circRNA durch Fusion von circRNA-Sequenz- und Netzwerkinformationen
Warum winzige RNA-Schleifen und ihre Adressen wichtig sind
In jeder menschlichen Zelle sind unzählige RNA-Moleküle aktiv und steuern mit, welche Gene an- oder ausgeschaltet werden. Darunter finden sich die sogenannten circular RNAs oder circRNAs — ungewöhnliche, ringförmige RNA-Stücke, die überraschend stabil sind und mit vielen Erkrankungen, einschließlich Krebs, in Verbindung gebracht werden. Um zu verstehen, welche Funktion diese Moleküle übernehmen, brauchen Forschende eine grundlegende Information: wo sie sich innerhalb der Zelle befinden. Das kartieren der „Adressen“ von circRNAs mit herkömmlichen Labormethoden ist jedoch zeitaufwendig, teuer und unvollständig. Diese Studie stellt eine neue rechnergestützte Methode namens CircLoc vor, die vorhersagt, wo sich circRNAs in der Zelle aufhalten, indem sie Informationen aus ihren Sequenzen und aus den komplexen biologischen Netzwerken, an denen sie beteiligt sind, kombiniert.
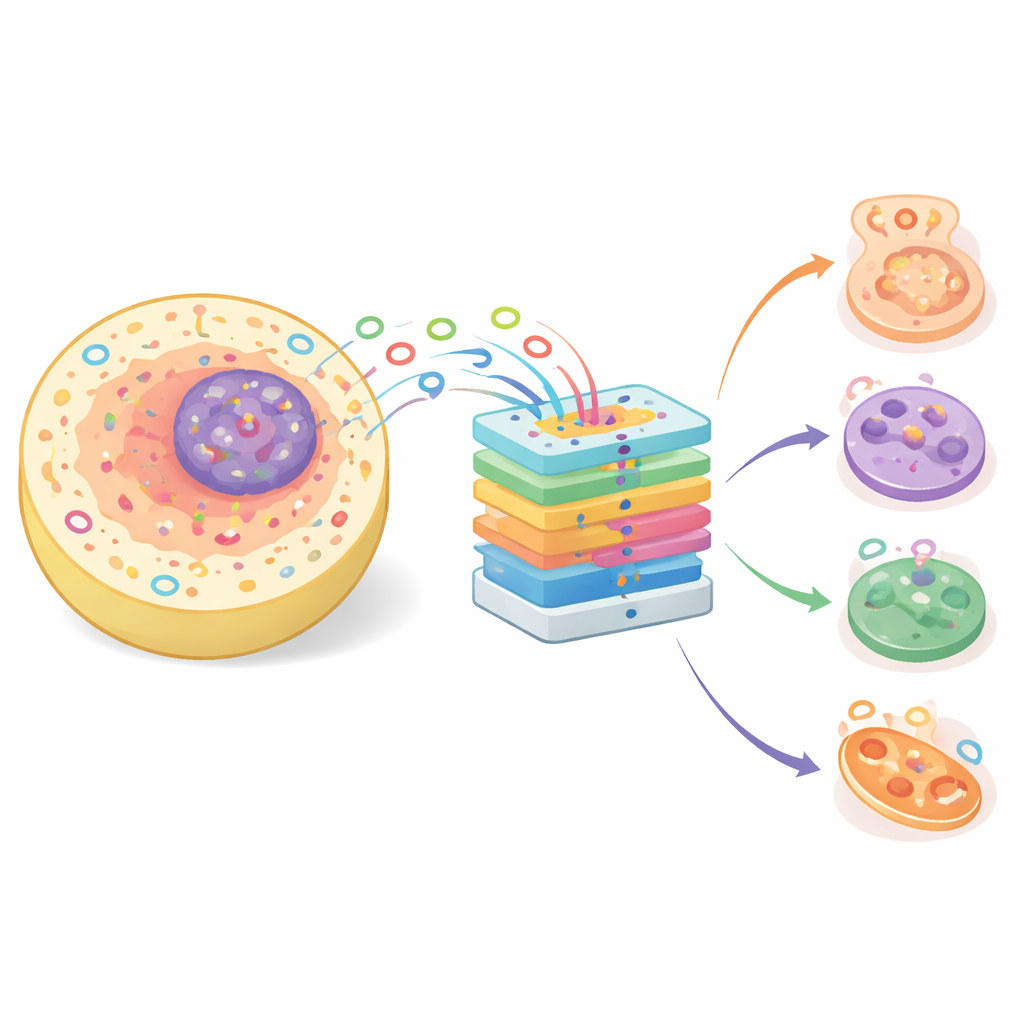
Kleine Schleifen mit großer biologischer Bedeutung
Früher als harmlose Nebenprodukte der Genverarbeitung abgetan, ist heute bekannt, dass circRNAs eine Reihe wichtiger Prozesse beeinflussen — von der Zelldifferenzierung bis zur Genregulation. Ihre kreisförmige Struktur macht sie stabiler als viele andere RNAs, was sie als potenzielle Biomarker zur Krankheitsdiagnose attraktiv macht. CircRNAs können Proteine binden und microRNAs — winzige Regulatoren, die normalerweise Genaktivität dämpfen — „auffangen“ und so das zelluläre Verhalten umgestalten. Da viele Moleküle nur in bestimmten Zellbereichen wirken, etwa im Zellkern, im Zytoplasma oder an Membranen, liefern Informationen zur subzellulären Lokalisation einer circRNA wichtige Hinweise auf ihre Funktion und ihre mögliche Rolle bei Gesundheit und Krankheit.
Aus verstreuten Daten ein Trainingsfeld machen
Die Autoren begonnen damit, eine sorgfältig kuratierte Sammlung menschlicher circRNAs mit bekannten Lokalisationen aus mehreren öffentlichen Datenbanken zusammenzustellen. Nach dem Entfernen seltener Kategorien und stark unausgewogener Gruppen konzentrierten sie sich auf sieben Hauptzellregionen, darunter Kern, Nukleolus, Nukleoplasma, Zytoplasma, Zytosol, Chromatin und Membranen. Insgesamt sammelten sie 1.486 circRNAs mit verlässlichen Sequenzdaten und mindestens einem bekannten Aufenthaltsort; viele gehörten gleichzeitig zu mehreren Regionen, was die Aufgabe zu einem echten Multi-Label-Vorhersageproblem machte. Zusätzliche Datensätze aus früheren Datenbankversionen und aus einer großen, krebsbezogenen Sammlung wurden als unabhängige Tests zurückgestellt, um zu prüfen, wie gut das Modell auf neu gemeldete circRNAs verallgemeinert.
Sequenzmuster mit zellulären Interaktionskarten verschmelzen
Die Kernidee von CircLoc ist, dass die „Adresse“ einer circRNA nicht nur durch ihre eigene Sequenz bestimmt wird, sondern auch durch ihr Umfeld. Auf der Sequenzseite betrachtet das Modell kurze Sequenzfragmente (K-mere und deren reverse Komplementärsequenzen) sowie reichhaltigere Muster, die ein großes, auf RNA spezialisiertes Sprachmodell namens RNAErnie gelernt hat; dieses wurde ursprünglich an umfangreichen RNA-Datensätzen trainiert, um subtile Regelmäßigkeiten zu erfassen. Auf der Netzwerkseite erstellten die Autoren mehrere Karten, die zeigen, wie circRNAs miteinander und mit verwandten biologischen Entitäten verbunden sind: überlappende Sequenzen, assoziierte Krankheiten, Arzneimittelreaktionen, interagierende microRNAs und bindende Proteine. Ein Netzwerk-Embedding-Werkzeug namens node2vec wandelt die Struktur jeder Karte in numerische Merkmale um, und ein Graph Attention Auto-Encoder (GATE) verfeinert diese dann, indem er Verbindungen zwischen ähnlich agierenden circRNAs betont und damit netzwerkbasierte Signale entrauscht und anreichert.
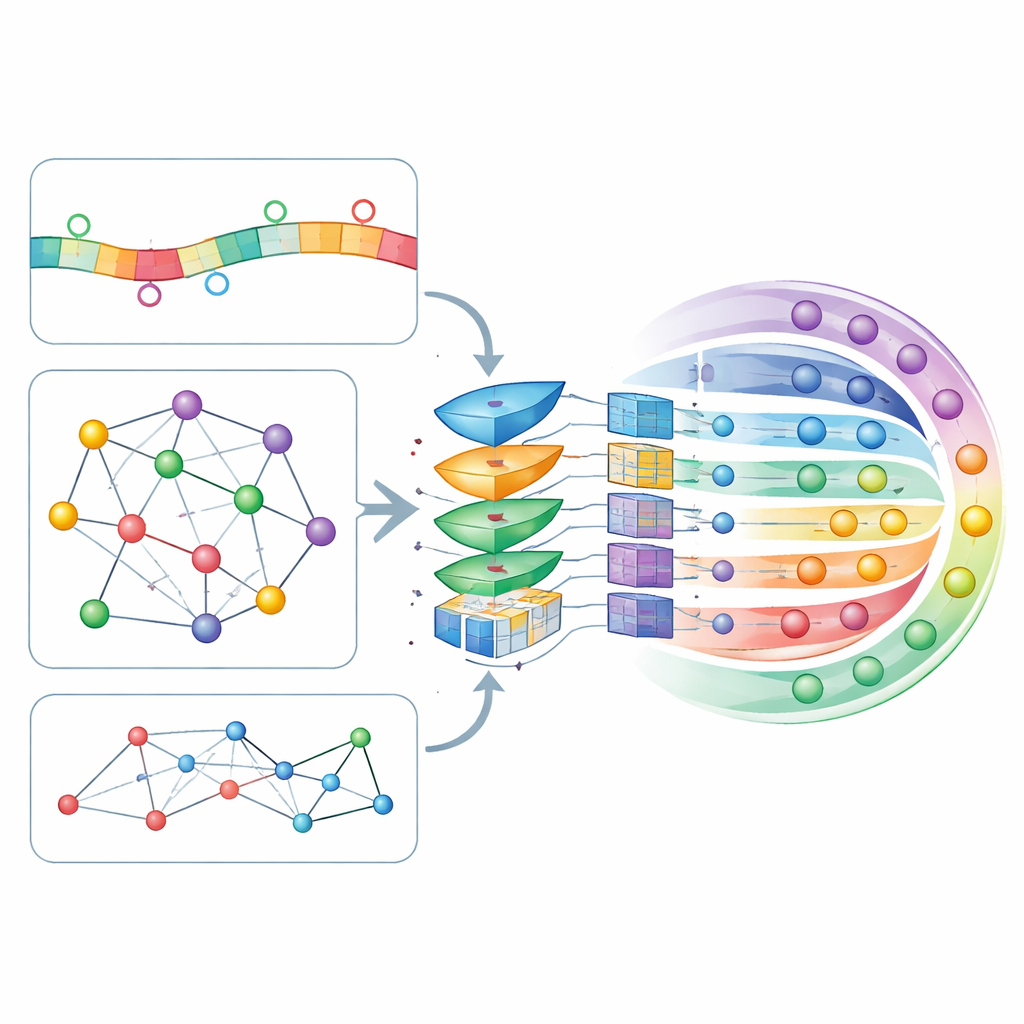
Das Modell entscheiden lassen, was am wichtigsten ist
All diese sequenz- und netzwerkbasierten Merkmale werden zu einem einzigen Profil pro circRNA zusammengeführt und durch eine Self-Attention-Schicht geleitet — ein Mechanismus, der dem Modell erlaubt zu lernen, welche Merkmal-Kombinationen den größten Einfluss auf seine Entscheidungen haben sollten. Die verfeinerten Profile werden anschließend in ein tiefes, voll verbundenes neuronales Netz eingespeist, das für jeden der sieben möglichen Aufenthaltsorte eine Wahrscheinlichkeit ausgibt. Die Autoren optimierten zahlreiche Einstellungen des Modells mittels zehnfacher Kreuzvalidierung, einem strengen Verfahren, das die Daten wiederholt in Trainings- und Testmengen aufteilt. CircLoc erreichte im Durchschnitt einen Wert von etwa 0,79 auf einer gängigen Qualitätsmetrik (AUC) und übertraf damit deutlich frühere Ansätze, die für microRNAs entwickelt wurden, sowie klassische Multi-Label-Methoden, die auf denselben Merkmalen trainiert wurden. Experimente, in denen bestimmte Merkmale oder Module entfernt wurden, zeigten, dass die Netzwerkdaten und die GATE-Verfeinerung besonders wichtig waren, während Sequenzmerkmale weiterhin nützliche, wenn auch kleinere, Verbesserungen beitrugen.
Wie gut kommt das Modell mit neuen circRNAs zurecht?
Um die praktische Nutzbarkeit zu prüfen, trainierte das Team CircLoc an einer Version der Lokalisationsdatenbank und testete es an circRNAs, die erst in einer späteren Version auftauchten, sowie an einer separaten, krebsfokussierten Ressource. Die Leistung sank gegenüber dem ursprünglichen Trainingssatz, was zu erwarten ist, wenn man echte neue Daten aus anderen Quellen betrachtet, blieb jedoch respektabel: Die durchschnittlichen Werte fielen moderat ab, zeigten aber weiterhin eine aussagekräftige Vorhersagekraft. Diese Tests zusammen mit Vergleichen zu anderen Methoden deuten darauf hin, dass CircLoc brauchbare Erstvorhersagen für die Lokalisation neu entdeckter circRNAs liefern kann — selbst wenn einige unterstützende Informationen, etwa zu Krankheiten oder Arzneimittelassoziationen, fehlen.
Was das für die zukünftige RNA-Forschung bedeutet
Diese Arbeit zeigt, dass die Kombination direkter Sequenzinformationen mit reichhaltigen Interaktionsnetzwerken Rechenmodelle dabei unterstützen kann, vorherzusagen, wo circRNAs innerhalb einer Zelle zu finden sind. Für experimentelle Biologen bietet CircLoc eine Möglichkeit, zu priorisieren, welche circRNAs in welchen Zellkompartimenten untersucht werden sollten, und kann so Zeit und Ressourcen sparen. Obwohl die Methode Laborbefunde noch nicht ersetzen kann und ihre Entwickler auf Einschränkungen wie unvollständige Daten und mäßige Leistung in einigen Tests hinweisen, stellt sie einen wichtigen Schritt hin zu groß angelegten, in silico erstellten „Adressbüchern“ für RNA-Moleküle dar. Mit wachsender Datenbasis und verbesserten Modellierungstechniken könnten solche Werkzeuge alltägliche Begleiter zu Experimenten werden und die Suche nach circRNAs leiten, die in Krankheit und Therapie besonders relevant sind.
Zitation: Chen, L., Hu, J. & Zhou, B. Predicting circRNA subcellular localization by fusing circRNA sequence and network information. Sci Rep 16, 12775 (2026). https://doi.org/10.1038/s41598-026-43808-x
Schlüsselwörter: kreisförmige RNA, subzelluläre Lokalisation, computationale Biologie, maschinelles Lernen, RNA-Netzwerke