Clear Sky Science · en
Predicting circRNA subcellular localization by fusing circRNA sequence and network information
Why tiny RNA loops and their addresses matter
Inside every human cell, vast numbers of RNA molecules bustle about, helping control which genes turn on and off. Among them are circular RNAs, or circRNAs—unusual loop-shaped pieces of RNA that are surprisingly stable and closely linked to many diseases, including cancers. To understand what these molecules do, scientists need to know a basic fact: where in the cell they live. Yet mapping the “addresses” of circRNAs with traditional lab experiments is slow, costly, and incomplete. This study presents a new computer-based method, called CircLoc, that predicts where circRNAs reside inside cells by combining information from their sequences and from the complex biological networks they participate in.
Small loops with big biological roles
Once dismissed as harmless leftovers of gene processing, circRNAs are now known to influence a range of vital processes, from how cells differentiate to how genes are regulated. Their circular shape makes them more stable than many other RNAs, which in turn makes them attractive as potential biomarkers for diagnosing disease. CircRNAs can bind proteins and soak up microRNAs—tiny regulators that normally dampen gene activity—thereby reshaping cellular behavior. Because many molecules act only in specific parts of the cell, such as the nucleus, cytoplasm, or membranes, knowing the subcellular location of a circRNA provides important clues to its function and its possible role in health and disease.
Turning scattered data into a training ground
The authors began by assembling a carefully curated collection of human circRNAs with known locations from several public databases. After removing rare categories and extremely imbalanced groups, they focused on seven major cellular regions, including the nucleus, nucleolus, nucleoplasm, cytoplasm, cytosol, chromatin, and membranes. In total, they gathered 1,486 circRNAs with reliable sequence information and at least one known location; many belonged to multiple regions at once, making the task a true multi-label prediction problem. Additional datasets from earlier database releases and from a large cancer-related collection were set aside as independent tests, allowing the team to examine how well their model would generalize to newly reported circRNAs.
Blending sequence patterns with cellular interaction maps
CircLoc’s core idea is that a circRNA’s address is shaped not only by its own sequence, but also by the company it keeps. On the sequence side, the model looks at short sequence fragments (k-mers and their reverse complements) and at richer patterns learned by a large RNA-focused language model called RNAErnie, which was originally trained on massive RNA datasets to capture subtle regularities. On the network side, the authors built several maps showing how circRNAs connect to each other and to related biological entities: overlapping sequences, associated diseases, drug responses, interacting microRNAs, and binding proteins. A network embedding tool called node2vec converts the structure of each map into numerical features, and a graph attention auto-encoder (GATE) then refines them by emphasizing connections between circRNAs that behave similarly, effectively denoising and enriching the network-derived signals.
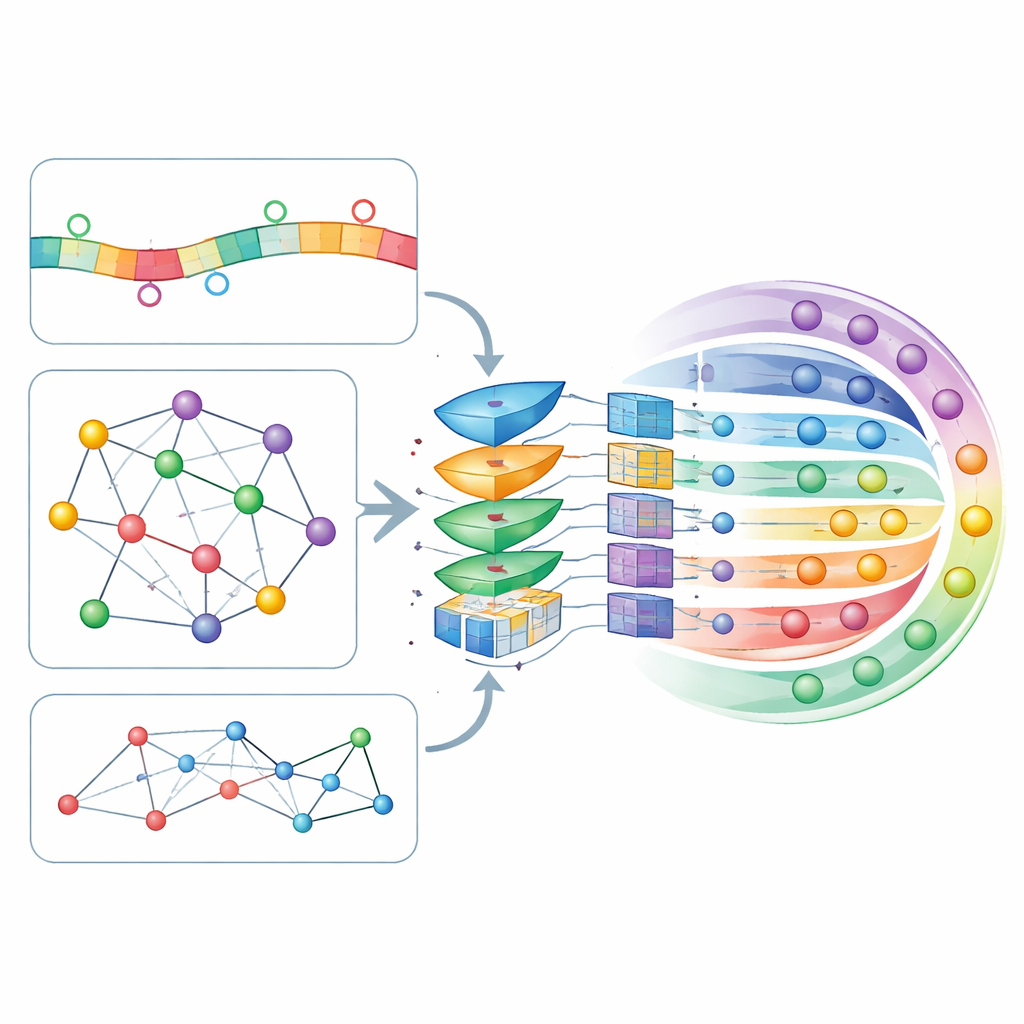
Letting the model decide what matters most
All these sequence- and network-based features are stitched together into a single profile for each circRNA and passed through a self-attention layer, a mechanism that lets the model learn which combinations of features should influence its decisions most strongly. The refined profiles then enter a deep, fully connected neural network that outputs a probability for each of the seven possible locations. The authors tuned the model’s many settings using ten-fold cross-validation, a rigorous procedure that repeatedly splits the data into training and testing portions. CircLoc achieved an average score of about 0.79 on a standard quality measure (AUC), clearly outperforming earlier approaches designed for microRNAs and classical multi-label methods trained on the same features. Experiments that removed specific features or modules showed that the network information and the GATE refinement were especially important, while sequence features still contributed useful, if smaller, improvements.
How well does the model handle new circRNAs?
To probe real-world usefulness, the team trained CircLoc on one version of the localization database and tested it on circRNAs that appeared only in a later release, as well as on a separate cancer-focused resource. Performance dipped compared with the original training set, as expected when facing genuinely new data from different sources, but remained respectable: average scores decreased modestly while still indicating meaningful predictive power. These tests, along with comparisons to other methods, suggest that CircLoc can provide reasonable first-pass guesses for the locations of newly discovered circRNAs, even when some supporting information—such as detailed disease or drug associations—is missing.
What this means for future RNA research
This work shows that combining direct sequence information with rich interaction networks can help computational models anticipate where circRNAs are likely to reside within a cell. For experimental biologists, CircLoc offers a way to prioritize which circRNAs to study in which cellular compartments, potentially saving time and resources. While the method cannot yet replace lab measurements, and its creators note limitations such as incomplete data and modest performance on some test sets, it represents an important step toward large-scale, in silico “address books” for RNA molecules. As databases grow and modeling techniques improve, such tools may become routine companions to experiments, guiding the search for circRNAs that matter most in disease and therapy.
Citation: Chen, L., Hu, J. & Zhou, B. Predicting circRNA subcellular localization by fusing circRNA sequence and network information. Sci Rep 16, 12775 (2026). https://doi.org/10.1038/s41598-026-43808-x
Keywords: circular RNA, subcellular localization, computational biology, machine learning, RNA networks