Clear Sky Science · sv
Djupinlärningsramverk med tolkbar features-selektion för exakt förutsägelse av SUMOylering
Varför detta är viktigt för hälsa och medicin
Proteiner driver nästan alla processer i våra celler, och små kemiska märken som läggs till efter att ett protein har bildats kan helt förändra dess funktion. Ett av dessa märken, kallat SUMO, har kopplats till cancer, Alzheimers sjukdom och andra allvarliga tillstånd. Att experimentellt fastställa exakt var SUMO fäster på tusentals proteiner är långsamt och kostsamt. Denna artikel presenterar Hybrid-Sumo, en kraftfull datorbaserad modell som kan peka ut sannolika SUMO-fästpunkter med imponerande noggrannhet, vilket potentiellt kan snabba på grundforskning och framtida läkemedelsupptäckt.
Hur celler växlar proteiners beteende
Våra celler finjusterar proteiners beteende genom ”efter-produktion”-ändringar kända som post-translationella modifieringar. SUMOylering är en sådan modifiering där ett litet modifierande protein fästs på en lysinbyggsten i ett målprotein. Denna lilla förändring kan påverka hur proteinet veckas, var det transporteras i cellen, hur länge det överlever och vilka partners det binder till. Eftersom SUMOylering påverkar genreglering, DNA-reparation och proteinåtervinning, är den starkt involverad i att upprätthålla cellulär balans. När SUMOylering går fel kan det bidra till neurodegenerativa sjukdomar och cancer, vilket gör noggranna kartor över SUMO-ställen till en hög prioritet för biologer.
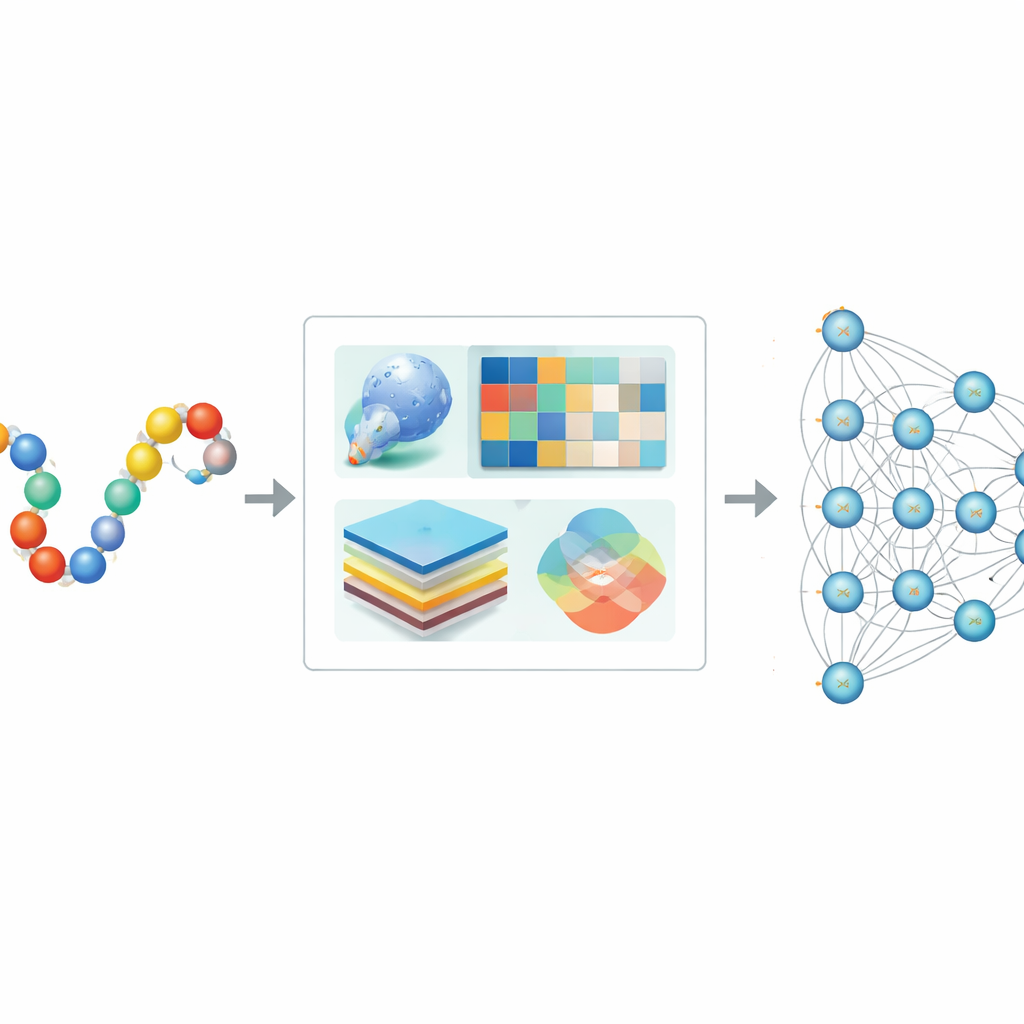
Varför datorer behövs för att hitta SUMO-märken
Traditionella laboratoriemetoder kan bekräfta SUMOylering, men de skalar inte väl till det enorma antalet proteiner i människor och andra organismer. Tidigare datorverktyg försökte upptäcka SUMO-ställen genom att känna igen korta sekvensmönster eller genom att använda klassiska maskininlärningsmetoder, såsom beslutsträd eller supportvektormaskiner. Dessa tillvägagångssätt var hjälpsamma men ignorerade ofta proteinets fulla kontext, inklusive dess tredimensionella form och evolutionära historia, och de hade svårt när data var obalanserade, med betydligt färre kända SUMO-ställen än icke-SUMO-ställen. Som ett resultat kunde förutsägelser bli snedvridna eller misslyckas med att generalisera till nya proteiner.
En hybrid syn på varje proteinplats
Författarna designade Hybrid-Sumo för att betrakta varje potentiellt SUMO-ställe från tre kompletterande vinklar samtidigt. Först använder de en strukturell måttstock kallad half-sphere exposure för att uppskatta hur nedgrävd eller exponerad en rest är på proteinets yta, vilket påverkar om SUMO fysiskt kan nå den. För det andra beräknar de evolutionära profiler som fångar hur en position förändrats över besläktade proteiner, och komprimerar dessa profiler med wavelet-metoder för att framhäva viktiga mönster samtidigt som brus reduceras. För det tredje lånar de en transformer-modell som ursprungligen byggts för språkbehandling och behandlar aminosyrasekvenser som meningar så att nätverket kan lära sig rika kontextuella ”inbäddningar” som beskriver hur varje position relaterar till sina grannar längs kedjan. Dessa tre uppsättningar siffror sammanfogas till en enda, detaljerad beskrivning av varje plats.
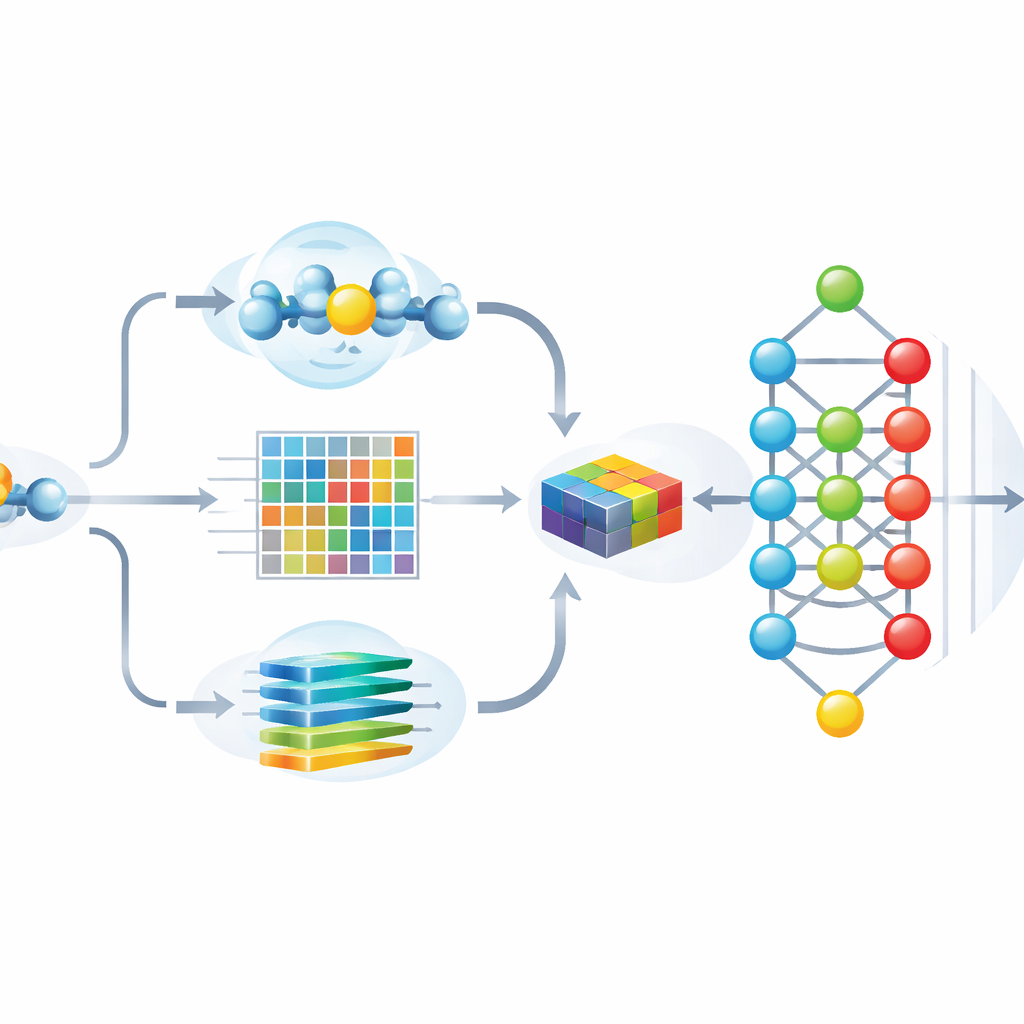
Göra modellen både exakt och förklarbar
Att mata in mer än tusen features i ett djupt neuralt nätverk kan förbättra noggrannheten, men det ökar också risken för överanpassning och gör det svårt att se vilka ledtrådar modellen verkligen förlitar sig på. För att hantera detta använder teamet en spelteori-inspirerad teknik kallad SHAP för att poängsätta hur mycket varje feature bidrar till korrekta förutsägelser. De behåller sedan endast de mest informativa 243 features, vilket dramatiskt minskar redundans samtidigt som prestandan bibehålls. Visuella analyser visar att efter denna selektion bildar SUMO- och icke-SUMO-ställen väl åtskilda kluster, och att de viktigaste features överensstämmer med intuitiva egenskaper såsom ytexponering, lokal laddning och karakteristiska sekvensmönster runt den modifierade lysinen.
Att testa Hybrid-Sumo
För att skydda mot missvisande resultat byggde forskarna omsorgsfullt balanserade och obalanserade dataset från en kurerad databas för proteinmodifieringar, tog bort nära-duplicerade sekvenser och utvärderade Hybrid-Sumo med upprepad korsvalidering samt helt oberoende testset. Den slutliga modellen nådde ungefär 99,7 % noggrannhet på träningsdata och omkring 96 % noggrannhet på osett material, något men konsekvent bättre än flera starka djupinlärnings- och ensemblemetoder som byggts specifikt för samma uppgift. Statistiska tester bekräftade att vinsterna från SHAP-baserad features-selektion är verkliga snarare än slumpmässiga, och jämförelser med andra vanliga algoritmer visade att fördelen kommer från de hybrida funktionerna och noggrann optimering, inte bara från valet av ett djupt nätverk.
Vad detta betyder framöver
För icke-specialister är huvudbudskapet att Hybrid-Sumo erbjuder ett mer pålitligt sätt att förutsäga var SUMO-märket kommer att fästa på ett protein, genom att kombinera 3D-struktur, evolutionära signaler och moderna sekvens-”språk”modeller. Genom att minska trial-and-error i laboratoriet kan det hjälpa forskare att prioritera experiment, utforska hur SUMOylering bidrar till sjukdom och så småningom vägleda terapier som riktar sig mot eller utnyttjar denna subtila proteinknapp. Samma designprinciper — att kombinera mångsidiga synsätt på en molekyl och sedan använda tolkbar features-selektion — skulle också kunna anpassas för att förutsäga många andra typer av proteinmodifieringar som ligger bakom hälsa och sjukdom.
Citering: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Nyckelord: SUMOylation, proteinmodifikation, djupinlärning, features-selektion, bioinformatik