Clear Sky Science · pl
Ramowa głębokiego uczenia z interpretowalnym wyborem cech do dokładnego przewidywania miejsc SUMOylacji
Dlaczego ma to znaczenie dla zdrowia i medycyny
Białka sterują niemal wszystkimi procesami w naszych komórkach, a drobne chemiczne znaczniki dodawane po zsyntezowaniu białka mogą całkowicie zmienić jego funkcję. Jeden z tych znaczników, nazywany SUMO, został powiązany z rakiem, chorobą Alzheimera i innymi poważnymi schorzeniami. Eksperymentalne ustalanie dokładnych miejsc przyłączenia SUMO w tysiącach białek jest powolne i kosztowne. W artykule zaprezentowano Hybrid-Sumo — wydajny model komputerowy, który potrafi precyzyjnie wskazywać prawdopodobne miejsca przyłączenia SUMO, co może przyspieszyć badania podstawowe i odkrywanie leków.
Jak komórki przełączają zachowanie białek
Nasze komórki dopracowują działanie białek za pomocą „poprodukcyjnych” poprawek zwanych modyfikacjami potranslacyjnymi. SUMOylacja jest jedną z takich modyfikacji, w której mały białkowy modyfikator przyłączany jest do reszty lizynowej w białku docelowym. Ta drobna zmiana może zmienić sposób fałdowania białka, jego lokalizację w komórce, czas przeżycia i partnerów, z którymi się wiąże. Ponieważ SUMOylacja wpływa na kontrolę genów, naprawę DNA i recykling białek, odgrywa kluczową rolę w utrzymaniu równowagi komórkowej. Gdy proces ten zostaje zaburzony, może przyczyniać się do chorób neurodegeneracyjnych i nowotworów, co czyni tworzenie dokładnych map miejsc SUMO priorytetem dla biologów.
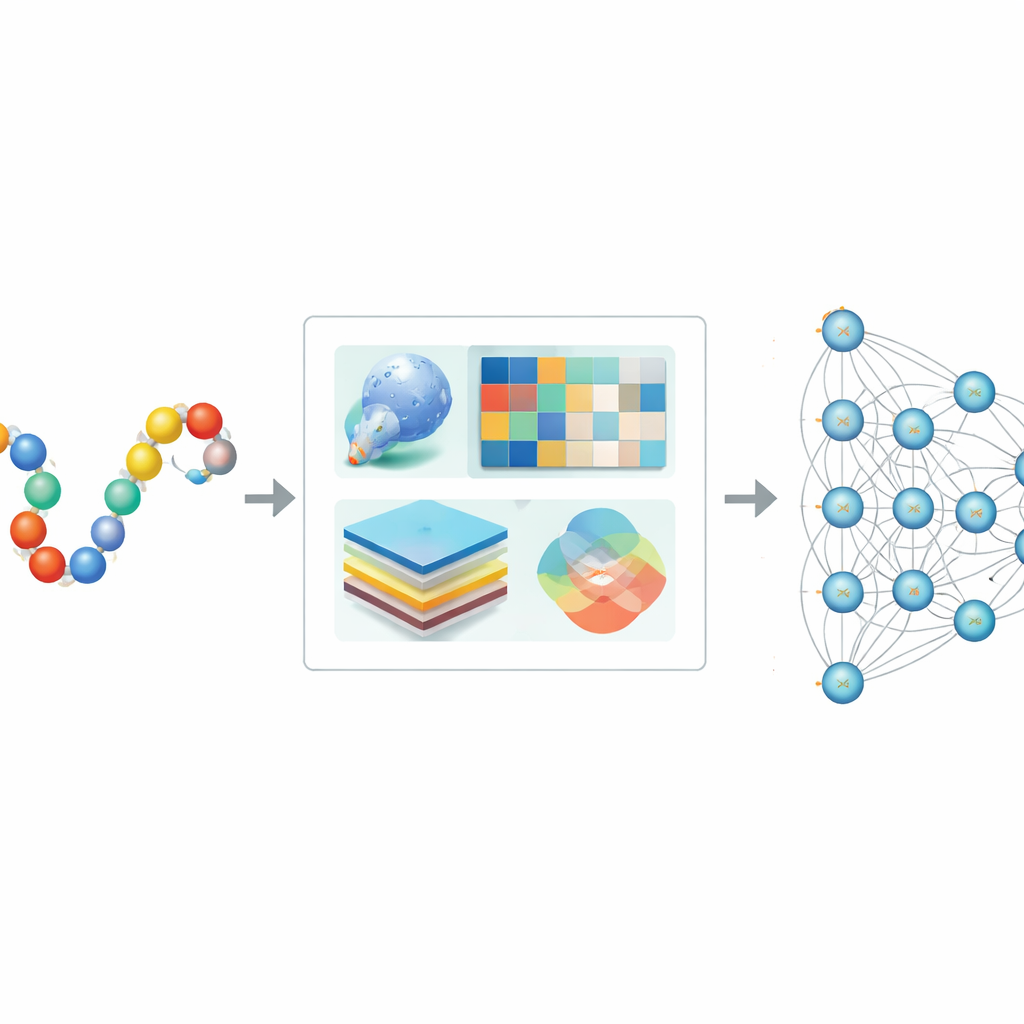
Dlaczego potrzebne są komputery do wykrywania znaczników SUMO
Tradycyjne metody laboratoryjne potwierdzają SUMOylację, ale nie skalują się dobrze do ogromnej liczby białek u ludzi i innych organizmów. Wcześniejsze narzędzia komputerowe próbowały wykrywać miejsca SUMO, rozpoznając krótkie wzorce sekwencji lub stosując klasyczne metody uczenia maszynowego, takie jak drzewa decyzyjne czy maszyny wektorów nośnych. Choć użyteczne, podejścia te często pomijały pełny kontekst białka, w tym jego trójwymiarową strukturę i historię ewolucyjną, a także miały problemy z niezrównoważonymi danymi, gdzie znanych miejsc SUMO było znacznie mniej niż miejsc bez SUMO. W efekcie przewidywania mogły być obciążone błędem lub nie uogólniać się na nowe białka.
Hybrydowe spojrzenie na każde miejsce w białku
Autorzy zaprojektowali Hybrid-Sumo tak, by jednocześnie analizował każde potencjalne miejsce SUMO z trzech uzupełniających się perspektyw. Po pierwsze stosują miarę strukturalną zwaną ekspozycją półsferyczną (half-sphere exposure), aby oszacować, jak zakryta lub odsłonięta jest reszta na powierzchni białka, co wpływa na dostępność SUMO. Po drugie obliczają profile ewolucyjne pokazujące, jak dana pozycja zmieniała się w pokrewnych białkach, a następnie kompresują te profile metodami falkowymi, by wyróżnić istotne wzorce i zredukować szum. Po trzecie wykorzystują model transformera pierwotnie stworzony dla języka, traktując sekwencje aminokwasów jak zdania, dzięki czemu sieć uczy się bogatych kontekstowych „osadzających” (embeddings) opisujących relacje pozycji z jej sąsiadami w łańcuchu. Te trzy zestawy informacji łączone są w szczegółowy opis każdego miejsca.
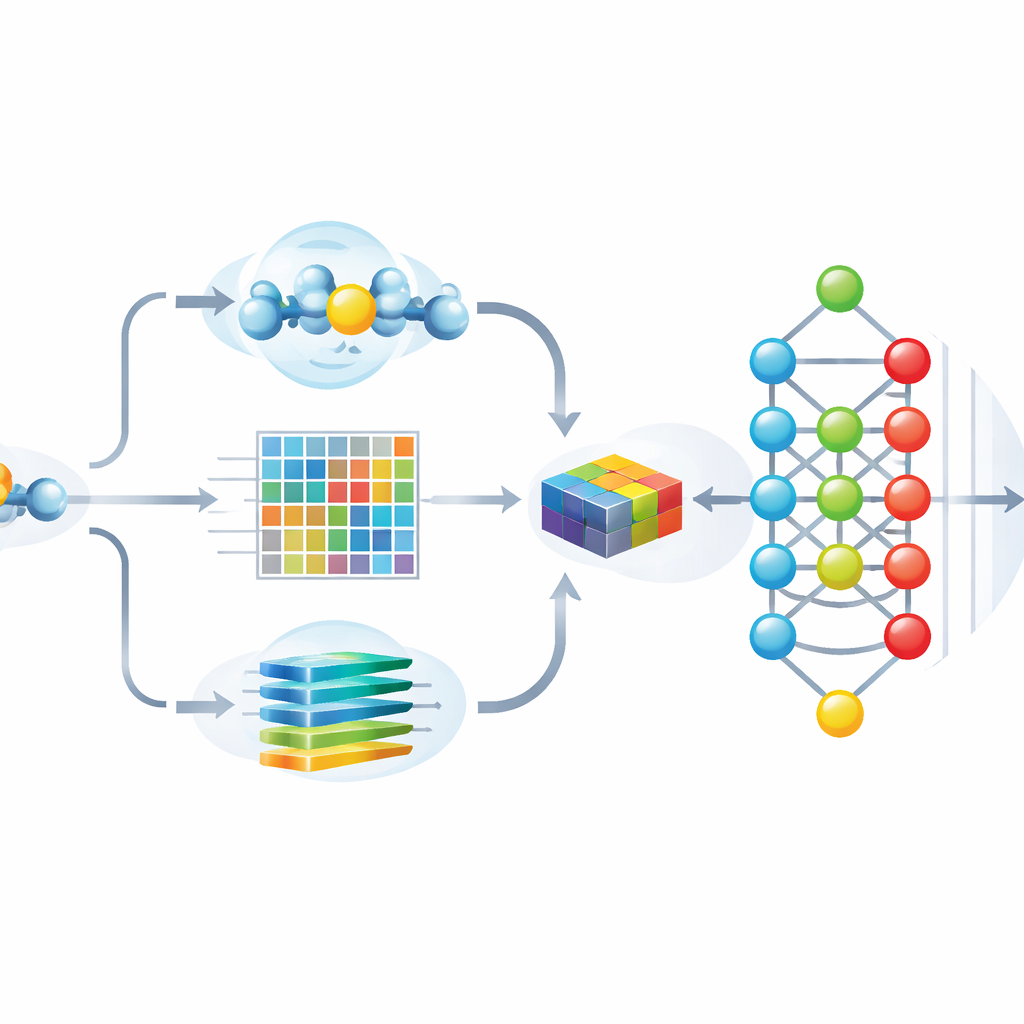
Uczynienie modelu zarówno dokładnym, jak i wyjaśnialnym
Podanie ponad tysiąca cech do głębokiej sieci neuronowej może poprawić dokładność, ale zwiększa też ryzyko przeuczenia i utrudnia zrozumienie, na których wnioskach model naprawdę się opiera. Aby temu zaradzić, zespół użył techniki inspirowanej teorią gier — SHAP — do oceny, jak bardzo każda cecha przyczynia się do poprawnych przewidywań. Następnie zachowali tylko najbardziej informacyjne 243 cechy, znacząco ograniczając redundancję przy zachowaniu wydajności. Analizy wizualne pokazują, że po tym wyborze miejsca SUMO i bez SUMO tworzą dobrze odseparowane klastry, a najważniejsze cechy zgodne są z intuicyjnymi właściwościami, takimi jak ekspozycja powierzchniowa, lokalny ładunek i charakterystyczne wzorce sekwencji wokół modyfikowanej lizyny.
Testowanie Hybrid-Sumo
Aby uniknąć mylących wyników, badacze starannie zbudowali zrównoważone i niezrównoważone zbiory danych z kuratorowanej bazy modyfikacji białek, usunęli niemal identyczne sekwencje i ocenili Hybrid-Sumo przy użyciu powtarzanego walidacyjnego podziału krzyżowego oraz całkowicie niezależnych zestawów testowych. Finalny model osiągnął około 99,7% dokładności na danych treningowych i około 96% na nieznanych białkach, nieznacznie, lecz konsekwentnie przewyższając kilka silnych metod głębokiego uczenia i metod zespołowych zaprojektowanych do tego samego zadania. Testy statystyczne potwierdziły, że korzyści z wyboru cech opartego na SHAP są istotne, a porównania z innymi powszechnymi algorytmami wykazały, że przewaga wynika z hybrydowych cech i starannej optymalizacji, a nie wyłącznie z zastosowania sieci głębokiej.
Co to oznacza na przyszłość
Dla odbiorców niebędących specjalistami kluczowym przesłaniem jest to, że Hybrid-Sumo oferuje bardziej wiarygodny sposób przewidywania, gdzie znacznik SUMO przyłączy się do białka, łącząc informacje o strukturze 3D, sygnały ewolucyjne i nowoczesne modele „języka” sekwencji. Ograniczając metodę prób i błędów w laboratorium, może pomóc naukowcom w priorytetyzowaniu eksperymentów, badaniu roli SUMOylacji w chorobach i w końcu w projektowaniu terapii celujących w ten subtelny przełącznik białkowy. Te same zasady projektowe — łączenie różnych spojrzeń na cząsteczkę i stosowanie interpretowalnego wyboru cech — można również dostosować do przewidywania wielu innych rodzajów modyfikacji białek leżących u podstaw zdrowia i chorób.
Cytowanie: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Słowa kluczowe: SUMOylacja, modyfikacja białka, głębokie uczenie, wybór cech, bioinformatyka