Clear Sky Science · de
Tiefes Lernframework mit interpretierbarer Merkmalsauswahl zur genauen Vorhersage von SUMOylierungsstellen
Warum das für Gesundheit und Medizin wichtig ist
Proteine steuern nahezu jeden Prozess in unseren Zellen, und winzige chemische Anhänge, die nach der Proteinsynthese hinzugefügt werden, können deren Funktion grundlegend verändern. Einer dieser Anhänge, SUMO genannt, wird mit Krebs, Alzheimer und anderen schweren Erkrankungen in Verbindung gebracht. Experimentell genau festzustellen, wo SUMO an Tausenden von Proteinen ansetzt, ist zeitaufwendig und teuer. Dieser Artikel stellt Hybrid-Sumo vor, ein leistungsfähiges Computermodell, das wahrscheinliche SUMO-Bindungsstellen mit beeindruckender Genauigkeit identifizieren kann und so Grundlagenforschung und zukünftige Wirkstoffforschung beschleunigen könnte.
Wie Zellen das Verhalten von Proteinen steuern
Unsere Zellen justieren das Verhalten von Proteinen durch „nachgelagerte“ Veränderungen, bekannt als posttranslationale Modifikationen. SUMOylierung ist eine solche Modifikation, bei der ein kleines Modifikatorprotein an ein Lysin in einem Zielprotein angefügt wird. Diese kleine Änderung kann beeinflussen, wie das Protein faltet, wohin es in der Zelle transportiert wird, wie lange es überdauert und mit welchen Partnern es interagiert. Da SUMOylierung die Genregulation, die DNA-Reparatur und das Proteinrecycling beeinflusst, spielt sie eine zentrale Rolle beim Erhalt des zellulären Gleichgewichts. Fehlregulation der SUMOylierung kann zu neurodegenerativen Erkrankungen und Krebs beitragen, weshalb genaue Karten von SUMO-Stellen für Biologen hohe Priorität haben.
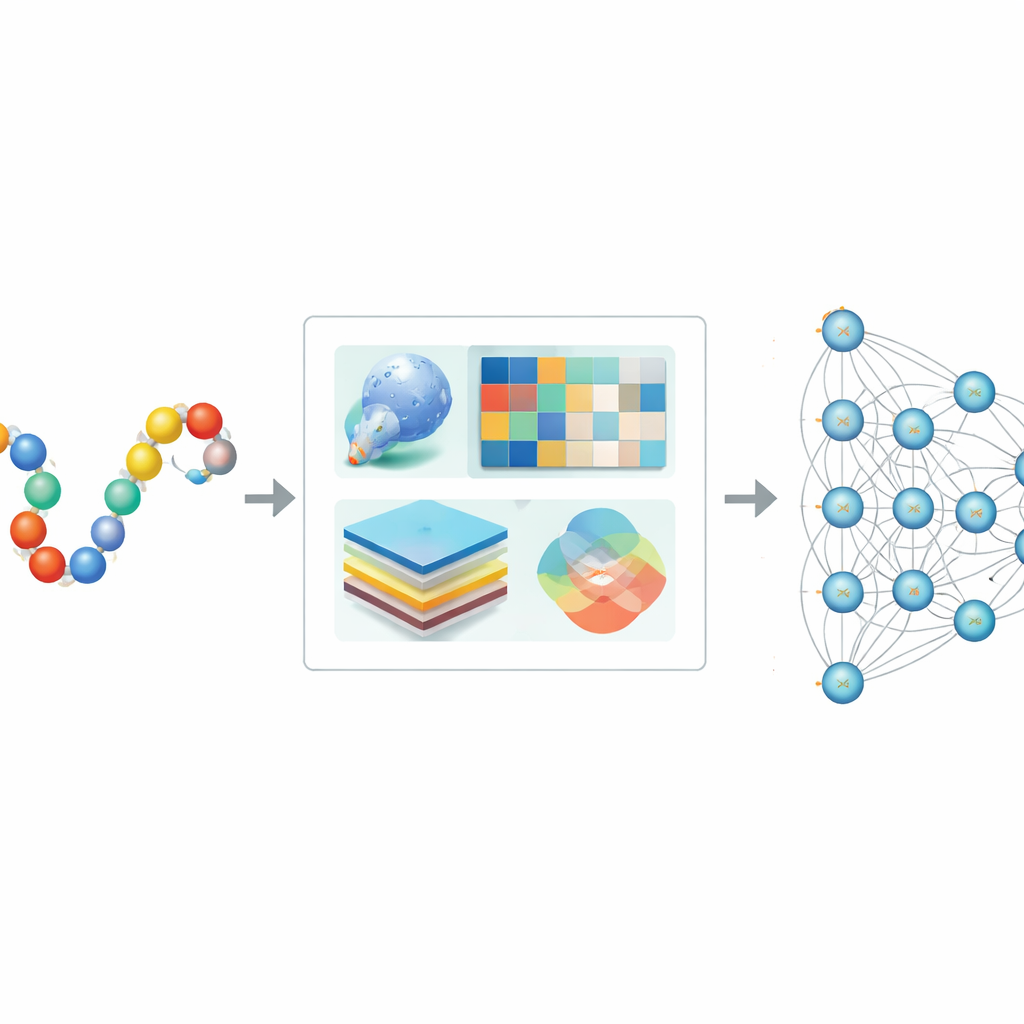
Warum Computer nötig sind, um SUMO-Anhänge zu finden
Traditionelle Labormethoden können SUMOylierung bestätigen, skalierten jedoch nicht gut auf die enorme Anzahl von Proteinen beim Menschen und in anderen Organismen. Frühere computergestützte Werkzeuge versuchten, SUMO-Stellen anhand kurzer Sequenzmuster oder klassischer Machine-Learning-Methoden wie Entscheidungsbäumen oder Support-Vektor-Maschinen zu erkennen. Diese Ansätze waren zwar nützlich, ließen jedoch oft den vollständigen Kontext eines Proteins außer Acht, darunter seine dreidimensionale Struktur und evolutionäre Geschichte, und hatten mit unausgewogenen Daten zu kämpfen, bei denen bekannte SUMO-Stellen deutlich seltener sind als Nicht-SUMO-Stellen. Infolgedessen konnten Vorhersagen verzerrt sein oder bei neuen Proteinen nicht generalisieren.
Eine hybride Sicht auf jede Proteinstelle
Die Autoren entwickelten Hybrid-Sumo, um jede potenzielle SUMO-Stelle gleichzeitig aus drei komplementären Blickwinkeln zu betrachten. Erstens verwenden sie ein strukturelles Maß namens Half-Sphere Exposure, um abzuschätzen, wie eingebettet oder exponiert eine Aminosäure auf der Proteinoberfläche ist — ein Faktor dafür, ob SUMO physisch zugänglich ist. Zweitens berechnen sie evolutionäre Profile, die zeigen, wie sich eine Position in verwandten Proteinen verändert hat, und komprimieren diese Profile mit Wavelet-Methoden, um wichtige Muster hervorzuheben und Rauschen zu reduzieren. Drittens nutzen sie ein Transformer-Modell, ursprünglich für Sprache entwickelt, und behandeln Aminosäuresequenzen wie Sätze, sodass das Netzwerk reichhaltige kontextuelle „Embeddings“ lernen kann, die beschreiben, wie jede Position zu ihren Nachbarn in der Kette steht. Diese drei Merkmalsgruppen werden zu einer einzigen, detaillierten Beschreibung jeder Stelle verschmolzen.
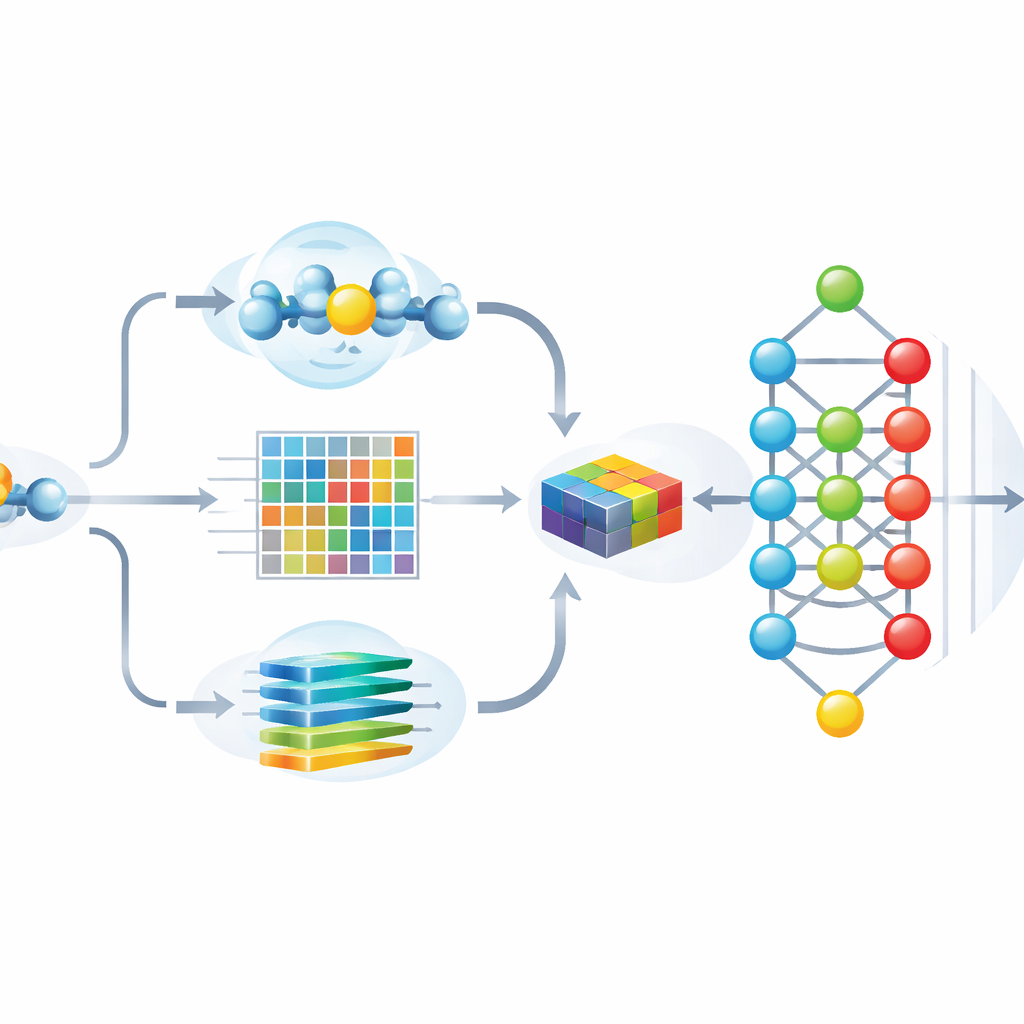
Das Modell sowohl genau als auch erklärbar machen
Mehr als tausend Merkmale in ein tiefes neuronales Netzwerk einzuspeisen kann die Genauigkeit erhöhen, birgt aber das Risiko von Überanpassung und erschwert es, nachzuvollziehen, auf welche Hinweise das Modell tatsächlich setzt. Um dem zu begegnen, nutzt das Team eine spieltheorie-inspirierte Technik namens SHAP, um zu bewerten, wie stark jedes Merkmal zu korrekten Vorhersagen beiträgt. Anschließend behalten sie nur die informativsten 243 Merkmale bei, wodurch Redundanz drastisch reduziert wird, während die Leistung erhalten bleibt. Visuelle Analysen zeigen, dass nach dieser Auswahl SUMO- und Nicht-SUMO-Stellen gut getrennte Cluster bilden und die wichtigsten Merkmale mit plausiblen Eigenschaften übereinstimmen, wie Oberflächenexposition, lokale Ladung und charakteristische Sequenzmuster um das modifizierte Lysin.
Hybrid-Sumo auf die Probe gestellt
Um irreführende Ergebnisse zu vermeiden, bauten die Forscher sorgfältig balancierte und unbalancierte Datensätze aus einer kuratierten Proteinmodifikationsdatenbank, entfernten nahezu identische Sequenzen und bewerteten Hybrid-Sumo mittels wiederholter Kreuzvalidierung sowie vollständig unabhängiger Testsätze. Das finale Modell erreichte etwa 99,7 % Genauigkeit auf Trainingsdaten und rund 96 % Genauigkeit auf bisher ungesehenen Proteinen und übertraf damit leicht, aber konsistent mehrere starke Deep-Learning- und Ensemble-Methoden, die speziell für dieselbe Aufgabe entwickelt wurden. Statistische Tests bestätigten, dass die Verbesserungen durch die SHAP-basierte Merkmalsauswahl echt sind und nicht zufällig, und Vergleiche mit anderen gängigen Algorithmen zeigten, dass der Vorteil aus den hybriden Merkmalen und der sorgfältigen Optimierung stammt, nicht nur aus der Wahl eines tiefen Netzwerks.
Was das für die Zukunft bedeutet
Für Nicht-Fachleute ist die Kernbotschaft, dass Hybrid-Sumo eine verlässlichere Methode bietet, um vorherzusagen, wo der SUMO-Anhang an einem Protein landen wird, indem 3D-Struktur, evolutionäre Signale und moderne Sequenz-„Sprach“-Modelle kombiniert werden. Durch die Verringerung von Versuch-und-Irrtum im Labor kann es Wissenschaftlern helfen, Experimente zu priorisieren, zu erforschen, wie SUMOylierung zu Krankheiten beiträgt, und schließlich Therapien zu leiten, die diesen subtilen Proteinschalter ansprechen oder nutzen. Dieselben Gestaltungsprinzipien — das Kombinieren vielfältiger Blickwinkel auf ein Molekül und danach eine interpretierbare Merkmalsauswahl — könnten auch angepasst werden, um viele andere Arten von Proteinmodifikationen vorherzusagen, die Gesundheit und Krankheit zugrunde liegen.
Zitation: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Schlüsselwörter: SUMOylierung, Proteinmodifikation, Tiefes Lernen, Merkmalsauswahl, Bioinformatik