Clear Sky Science · es
Marco de aprendizaje profundo con selección de características interpretable para la predicción precisa de sitios de SUMOilación
Por qué esto importa para la salud y la medicina
Las proteínas regulan casi todos los procesos en nuestras células, y pequeñas etiquetas químicas añadidas después de la síntesis de una proteína pueden cambiar por completo su función. Una de estas etiquetas, denominada SUMO, se ha relacionado con el cáncer, la enfermedad de Alzheimer y otras dolencias graves. Identificar experimentalmente en qué puntos exactos se une SUMO en miles de proteínas es lento y costoso. Este artículo presenta Hybrid-Sumo, un potente modelo informático que puede localizar con notable precisión los sitios probables de unión de SUMO, acelerando potencialmente la investigación básica y el descubrimiento de fármacos en el futuro.
Cómo las células cambian el comportamiento de las proteínas
Nuestras células afinan el comportamiento de las proteínas mediante ediciones “posproducción” conocidas como modificaciones postraduccionales. La SUMOilación es una de estas modificaciones en la que una pequeña proteína modificadora se une a una lisina de la proteína objetivo. Este pequeño cambio puede alterar cómo se pliega la proteína, a qué compartimento celular se dirige, cuánto tiempo perdura y con qué socios interacciona. Dado que la SUMOilación influye en el control génico, la reparación del ADN y el reciclaje de proteínas, participa de forma profunda en el mantenimiento del equilibrio celular. Cuando la SUMOilación falla, puede contribuir a enfermedades neurodegenerativas y al cáncer, por lo que los mapas precisos de sitios de SUMO son una prioridad para los biólogos.
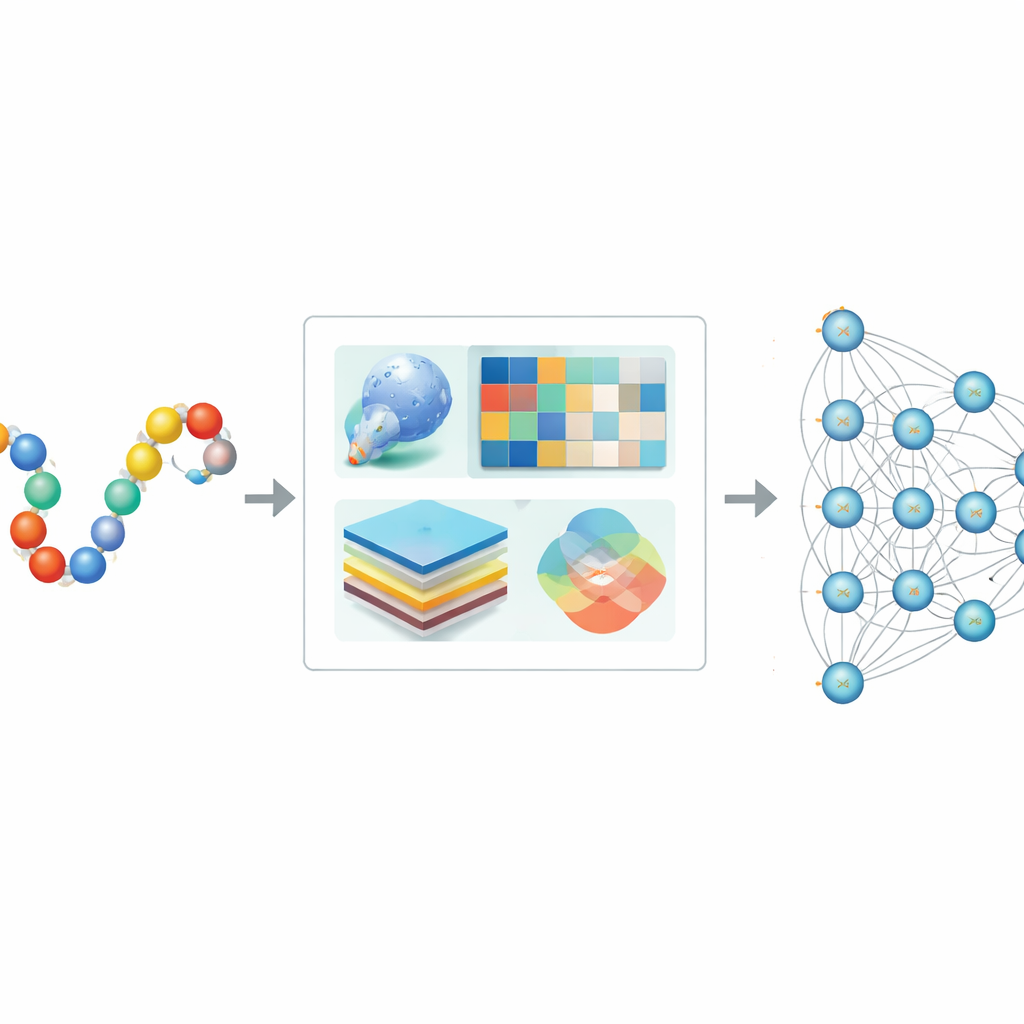
Por qué hacen falta ordenadores para encontrar etiquetas SUMO
Los métodos de laboratorio tradicionales pueden confirmar la SUMOilación, pero no escalan bien al enorme número de proteínas en humanos y otros organismos. Herramientas informáticas anteriores intentaron detectar sitios SUMO reconociendo patrones cortos de secuencia o usando métodos clásicos de aprendizaje automático, como árboles de decisión o máquinas de soporte vectorial. Aunque útiles, estos enfoques a menudo ignoraban el contexto completo de una proteína, incluida su forma tridimensional y su historia evolutiva, y tenían dificultades cuando los datos estaban desequilibrados, con muchos menos sitios SUMO conocidos que sitios no SUMO. Como resultado, las predicciones podían estar sesgadas o fallar al generalizar a nuevas proteínas.
Una visión híbrida de cada sitio proteico
Los autores diseñaron Hybrid-Sumo para examinar cada posible sitio de SUMO desde tres ángulos complementarios a la vez. Primero, usan una medida estructural llamada exposición de media esfera para estimar cuánto está enterrada o expuesta una residuo en la superficie de la proteína, lo que afecta si SUMO puede acceder físicamente a él. En segundo lugar, calculan perfiles evolutivos que capturan cómo ha cambiado una posición a través de proteínas relacionadas, y luego comprimen esos perfiles usando métodos wavelet para resaltar patrones importantes y reducir el ruido. En tercer lugar, toman prestado un modelo transformer originalmente creado para el lenguaje, tratando las secuencias de aminoácidos como oraciones para que la red aprenda “embeddings” contextuales ricos que describen cómo cada posición se relaciona con sus vecinas a lo largo de la cadena. Estos tres conjuntos de valores se fusionan en una única descripción detallada de cada sitio.
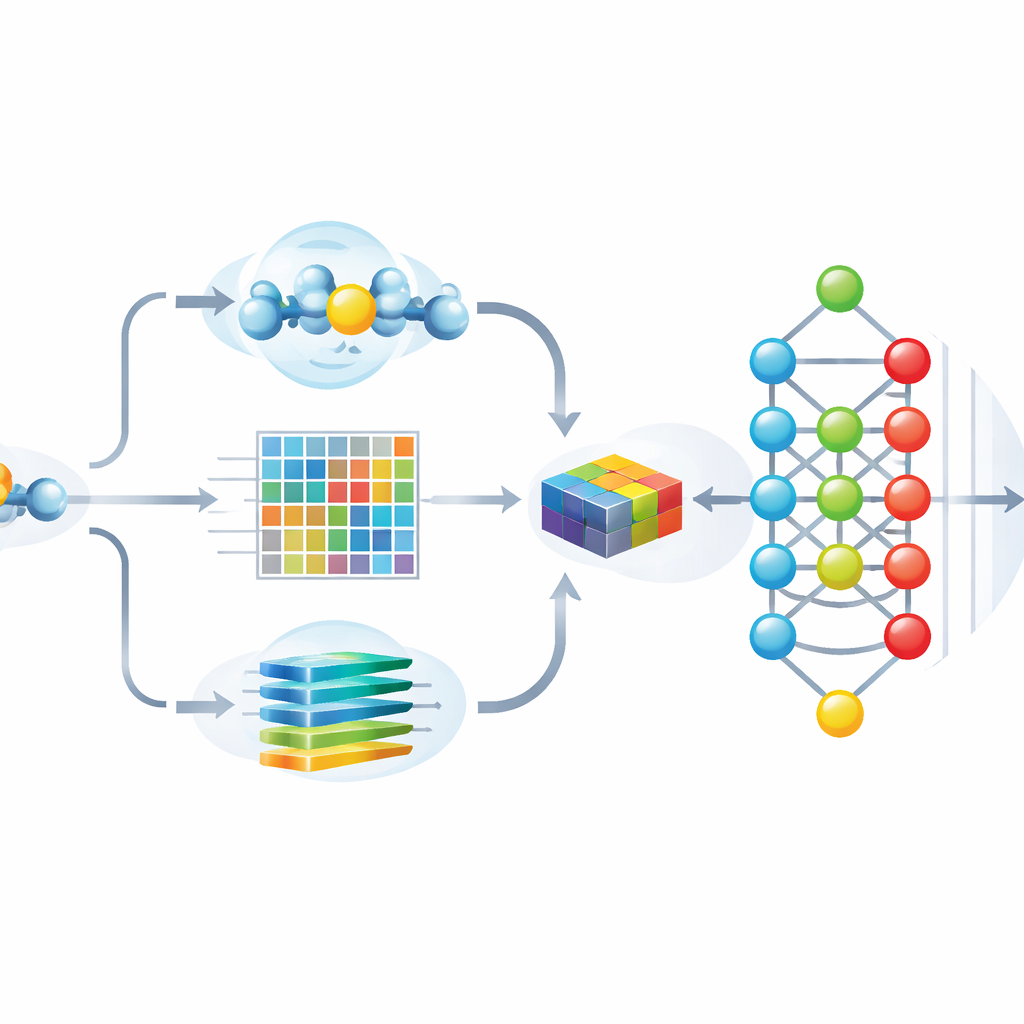
Hacer el modelo preciso y explicable
Alimentar más de mil características a una red neuronal profunda puede mejorar la precisión, pero también aumenta el riesgo de sobreajuste y dificulta ver en qué pistas se apoya realmente el modelo. Para abordar esto, el equipo usa una técnica inspirada en la teoría de juegos llamada SHAP para puntuar cuánto contribuye cada característica a las predicciones correctas. Luego conservan sólo las 243 características más informativas, recortando drásticamente la redundancia mientras mantienen el rendimiento. Los análisis visuales muestran que, tras esta selección, los sitios SUMO y no SUMO forman clústeres bien separados, y que las características más importantes se alinean con propiedades intuitivas como la exposición superficial, la carga local y patrones de secuencia característicos alrededor de la lisina modificada.
Poniendo a prueba a Hybrid-Sumo
Para evitar resultados engañosos, los investigadores construyeron cuidadosamente conjuntos de datos equilibrados y desequilibrados a partir de una base de datos curada de modificaciones proteicas, eliminaron secuencias casi idénticas y evaluaron Hybrid-Sumo usando validación cruzada repetida así como conjuntos de prueba completamente independientes. El modelo final alcanzó alrededor del 99,7 % de precisión en los datos de entrenamiento y aproximadamente un 96 % de precisión en proteínas no vistas, superando de forma ligera pero consistente a varios métodos sólidos de aprendizaje profundo y ensamblado diseñados específicamente para la misma tarea. Pruebas estadísticas confirmaron que las mejoras debidas a la selección de características basada en SHAP son reales y no azarosas, y las comparaciones con otros algoritmos comunes mostraron que la ventaja proviene de las características híbridas y la optimización cuidadosa, no sólo de elegir una red profunda.
Qué significa esto de cara al futuro
Para el público no especializado, el mensaje clave es que Hybrid-Sumo ofrece una forma más fiable de predecir dónde se colocará la etiqueta SUMO en una proteína, usando una mezcla de estructura 3D, señales evolutivas y modelos modernos de “lenguaje” de secuencias. Al reducir la prueba y error en el laboratorio, puede ayudar a los científicos a priorizar experimentos, explorar cómo la SUMOilación contribuye a la enfermedad y orientar eventualmente terapias que apunten o exploten este sutil interruptor proteico. Los mismos principios de diseño —combinar visiones diversas de una molécula y luego usar una selección de características interpretable— podrían adaptarse también para predecir muchos otros tipos de modificaciones de proteínas que subyacen a la salud y la enfermedad.
Cita: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Palabras clave: SUMOilación, modificación de proteínas, aprendizaje profundo, selección de características, bioinformática