Clear Sky Science · it
Framework di deep learning con selezione di feature interpretabile per la predizione accurata dei siti di SUMOilazione
Perché è importante per la salute e la medicina
Le proteine controllano quasi tutti i processi nelle nostre cellule, e piccole etichette chimiche aggiunte dopo la sintesi di una proteina possono modificare radicalmente la sua funzione. Una di queste etichette, chiamata SUMO, è stata collegata al cancro, alla malattia di Alzheimer e ad altre condizioni gravi. Individuare sperimentalmente esattamente dove il SUMO si attacca su migliaia di proteine è lento e costoso. Questo articolo presenta Hybrid-Sumo, un potente modello computazionale che può identificare con notevole accuratezza i probabili siti di attacco del SUMO, accelerando potenzialmente la ricerca di base e la scoperta di farmaci futuri.
Come le cellule modificano il comportamento delle proteine
Le nostre cellule regolano finemente il comportamento proteico tramite modifiche post-traduzionali, ovvero «edit» avvenuti dopo la sintesi. La SUMOilazione è una di queste modifiche in cui una piccola proteina modificatrice viene legata a una lisina della proteina bersaglio. Questo piccolo cambiamento può influenzare il modo in cui la proteina si ripiega, dove si localizza nella cellula, la sua stabilità e i partner con cui interagisce. Poiché la SUMOilazione influenza il controllo genico, la riparazione del DNA e il riciclo proteico, è profondamente coinvolta nel mantenimento dell’omeostasi cellulare. Quando la SUMOilazione è alterata, può contribuire a malattie neurodegenerative e al cancro, rendendo la mappatura accurata dei siti di SUMO una priorità per i biologi.
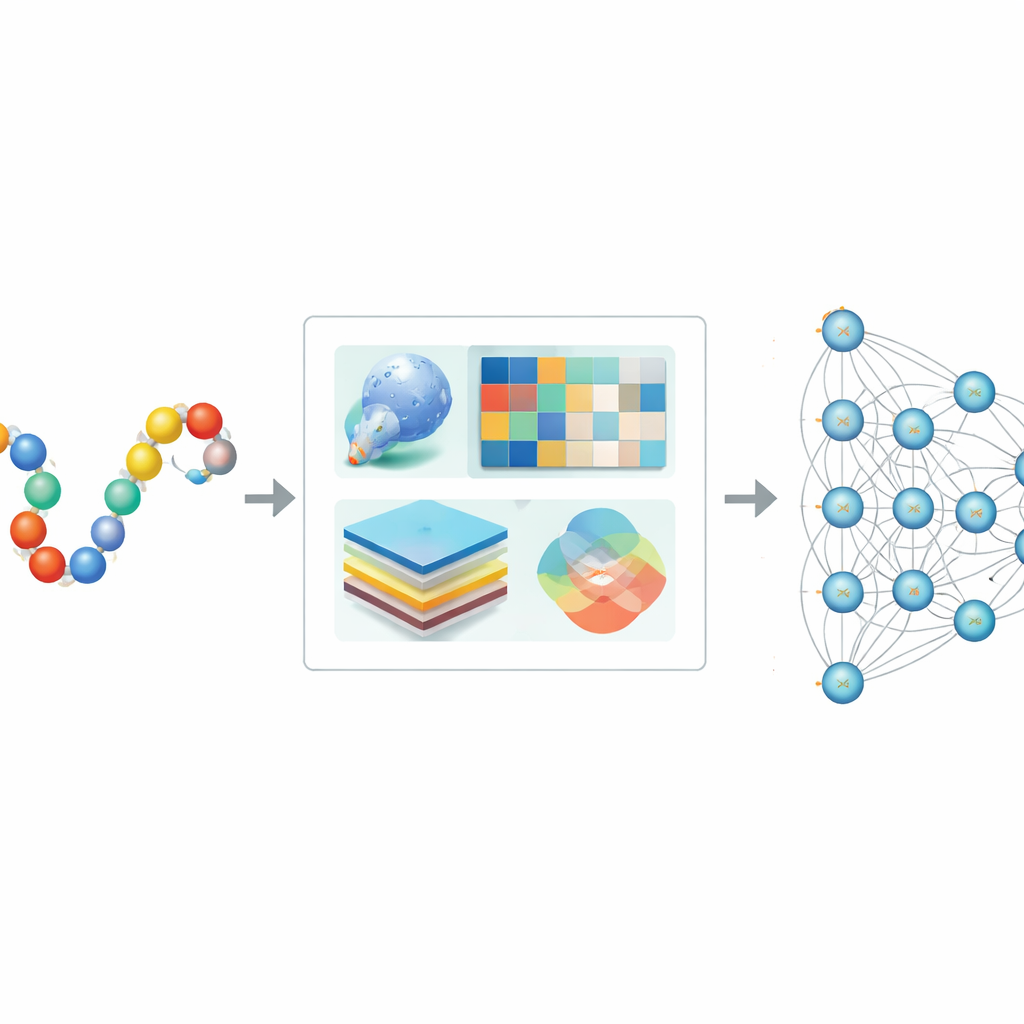
Perché servono i computer per trovare i tag SUMO
I metodi di laboratorio tradizionali possono confermare la SUMOilazione, ma non sono scalabili all’enorme numero di proteine negli esseri umani e in altri organismi. Strumenti computazionali precedenti cercavano di individuare i siti di SUMO riconoscendo brevi motivi di sequenza o usando metodi classici di machine learning, come gli alberi di decisione o le macchine a vettori di supporto. Pur essendo utili, questi approcci spesso ignoravano il contesto completo di una proteina, inclusa la sua struttura tridimensionale e la storia evolutiva, e faticavano quando i dati erano squilibrati, con molti meno siti SUMO noti rispetto ai siti non SUMO. Di conseguenza, le predizioni potevano risultare distorte o non generalizzarsi a nuove proteine.
Una visione ibrida di ogni sito proteico
Gli autori hanno progettato Hybrid-Sumo per analizzare ogni potenziale sito di SUMO da tre angolazioni complementari contemporaneamente. Innanzitutto, utilizzano una misura strutturale chiamata half-sphere exposure per stimare quanto un residuo sia sepolto o esposto sulla superficie della proteina, elemento che condiziona l’accessibilità fisica del SUMO. In secondo luogo, calcolano profili evolutivi che catturano come una posizione è variata tra proteine correlate, quindi comprimono questi profili usando metodi wavelet per evidenziare pattern importanti riducendo il rumore. Terzo, sfruttano un modello transformer originariamente sviluppato per il linguaggio, trattando le sequenze di amminoacidi come frasi in modo che la rete apprenda ricche ‘incorporazioni’ contestuali che descrivono come ogni posizione si rapporta ai vicini lungo la catena. Questi tre insiemi di dati vengono fusi in una singola descrizione dettagliata di ciascun sito.
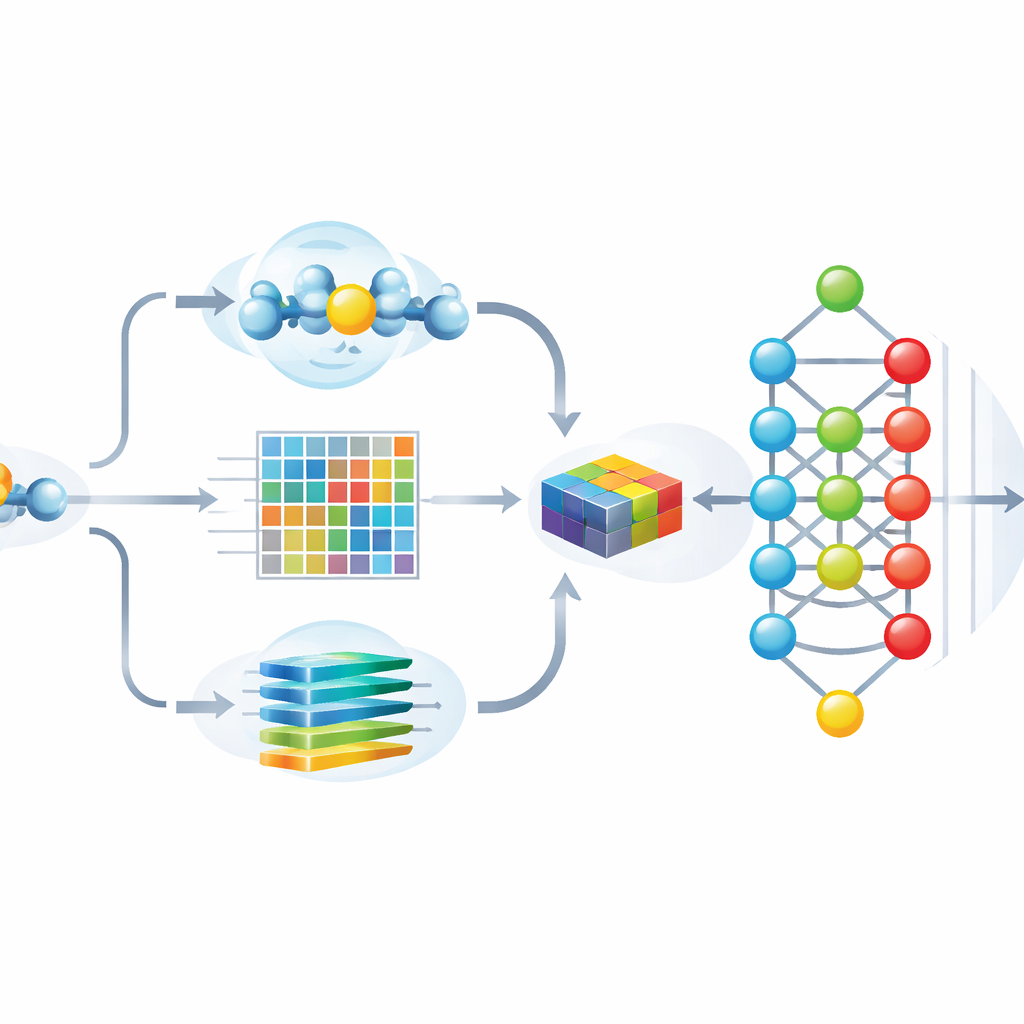
Resa del modello sia accurata sia interpretabile
Alimentare una rete neurale profonda con più di mille feature può migliorare l’accuratezza, ma aumenta anche il rischio di overfitting e rende difficile capire su quali indizi il modello si basi realmente. Per affrontare questo problema, il team usa una tecnica ispirata alla teoria dei giochi chiamata SHAP per valutare quanto ciascuna feature contribuisca alle predizioni corrette. Mantengono quindi solo le 243 feature più informative, riducendo drasticamente la ridondanza pur preservando le prestazioni. Analisi visive mostrano che, dopo questa selezione, i siti SUMO e non-SUMO formano cluster ben separati, e le feature più importanti si allineano a proprietà intuitive come l’esposizione superficiale, la carica locale e pattern di sequenza caratteristici attorno alla lisina modificata.
Testare Hybrid-Sumo
Per evitare risultati fuorvianti, i ricercatori hanno costruito con cura dataset bilanciati e sbilanciati da un database curato di modificazioni proteiche, rimosso sequenze quasi duplicate e valutato Hybrid-Sumo usando cross-validazione ripetuta oltre a set di test completamente indipendenti. Il modello finale ha raggiunto circa il 99,7% di accuratezza sui dati di addestramento e intorno al 96% su proteine non viste, superando in modo lieve ma consistente diversi metodi profondi e ensemble forti sviluppati specificamente per lo stesso compito. Test statistici hanno confermato che i miglioramenti dovuti alla selezione di feature basata su SHAP sono reali e non dovuti al caso, e i confronti con altri algoritmi comuni hanno mostrato che il vantaggio deriva dalle feature ibride e dall’ottimizzazione accurata, non solo dalla scelta di una rete profonda.
Cosa significa per il futuro
Per i non specialisti, il messaggio principale è che Hybrid-Sumo offre un modo più affidabile per prevedere dove il tag SUMO si legherà su una proteina, usando una combinazione di struttura 3D, segnali evolutivi e modelli di “linguaggio” di sequenze moderni. Riducendo tentativi ed errori in laboratorio, può aiutare gli scienziati a dare priorità agli esperimenti, esplorare come la SUMOilazione contribuisca alle malattie e, in futuro, orientare terapie che prendano di mira o sfruttino questo sottile interruttore proteico. Gli stessi principi di progetto—combinare viste diverse di una molecola e poi applicare una selezione di feature interpretabile—potrebbero essere adattati per prevedere molti altri tipi di modificazioni proteiche rilevanti per la salute e la malattia.
Citazione: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Parole chiave: SUMOilazione, modificazione proteica, deep learning, selezione delle feature, bioinformatica