Clear Sky Science · en
Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction
Why this matters for health and medicine
Proteins run nearly every process in our cells, and tiny chemical tags added after a protein is made can completely change what it does. One of these tags, called SUMO, has been tied to cancer, Alzheimer’s disease, and other serious conditions. Experimentally finding exactly where SUMO attaches on thousands of proteins is slow and expensive. This paper introduces Hybrid-Sumo, a powerful computer model that can pinpoint likely SUMO attachment sites with striking accuracy, potentially speeding up basic research and future drug discovery.
How cells switch protein behavior
Our cells fine-tune protein behavior using “post-production” edits known as post-translational modifications. SUMOylation is one such edit in which a small modifier protein is fastened to a lysine building block on a target protein. This small change can alter how that protein folds, where it travels in the cell, how long it survives, and which partners it binds. Because SUMOylation influences gene control, DNA repair, and protein recycling, it is deeply involved in maintaining cellular balance. When SUMOylation goes wrong, it can contribute to neurodegenerative diseases and cancer, making accurate maps of SUMO sites a high priority for biologists.
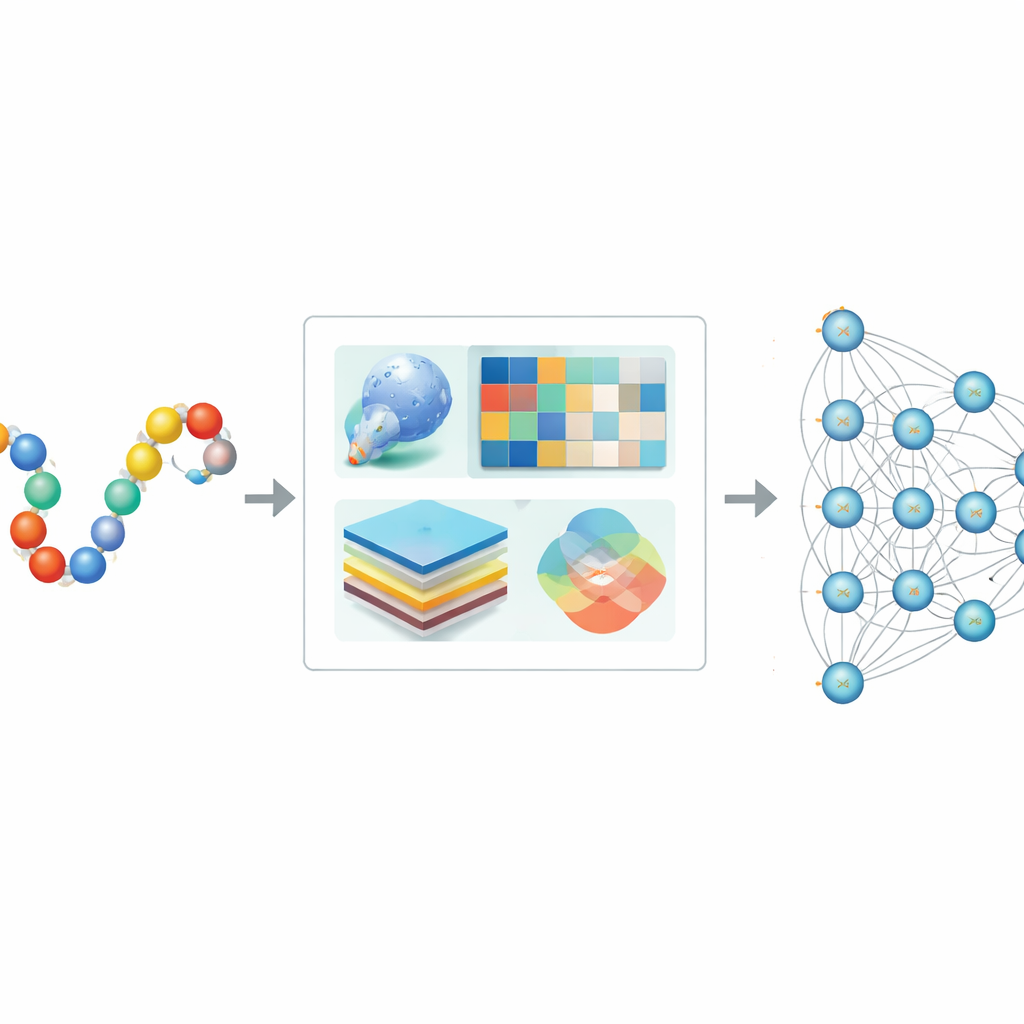
Why computers are needed to find SUMO tags
Traditional lab methods can confirm SUMOylation, but they do not scale well to the huge number of proteins in humans and other organisms. Earlier computer tools tried to spot SUMO sites by recognizing short sequence patterns or by using classic machine learning methods, such as decision trees or support vector machines. While helpful, these approaches often ignored the full context of a protein, including its three-dimensional shape and evolutionary history, and they struggled when the data were unbalanced, with far fewer known SUMO sites than non-SUMO sites. As a result, predictions could be biased or fail to generalize to new proteins.
A hybrid view of each protein site
The authors designed Hybrid-Sumo to look at each potential SUMO site from three complementary angles at once. First, they use a structural measure called half-sphere exposure to estimate how buried or exposed a residue is on the protein’s surface, which affects whether SUMO can physically reach it. Second, they compute evolutionary profiles that capture how a position has changed across related proteins, then compress these profiles using wavelet methods to highlight important patterns while reducing noise. Third, they borrow a transformer model originally built for language, treating amino acid sequences like sentences so the network can learn rich contextual “embeddings” that describe how each position relates to its neighbors along the chain. These three sets of numbers are fused into a single, detailed description of each site.
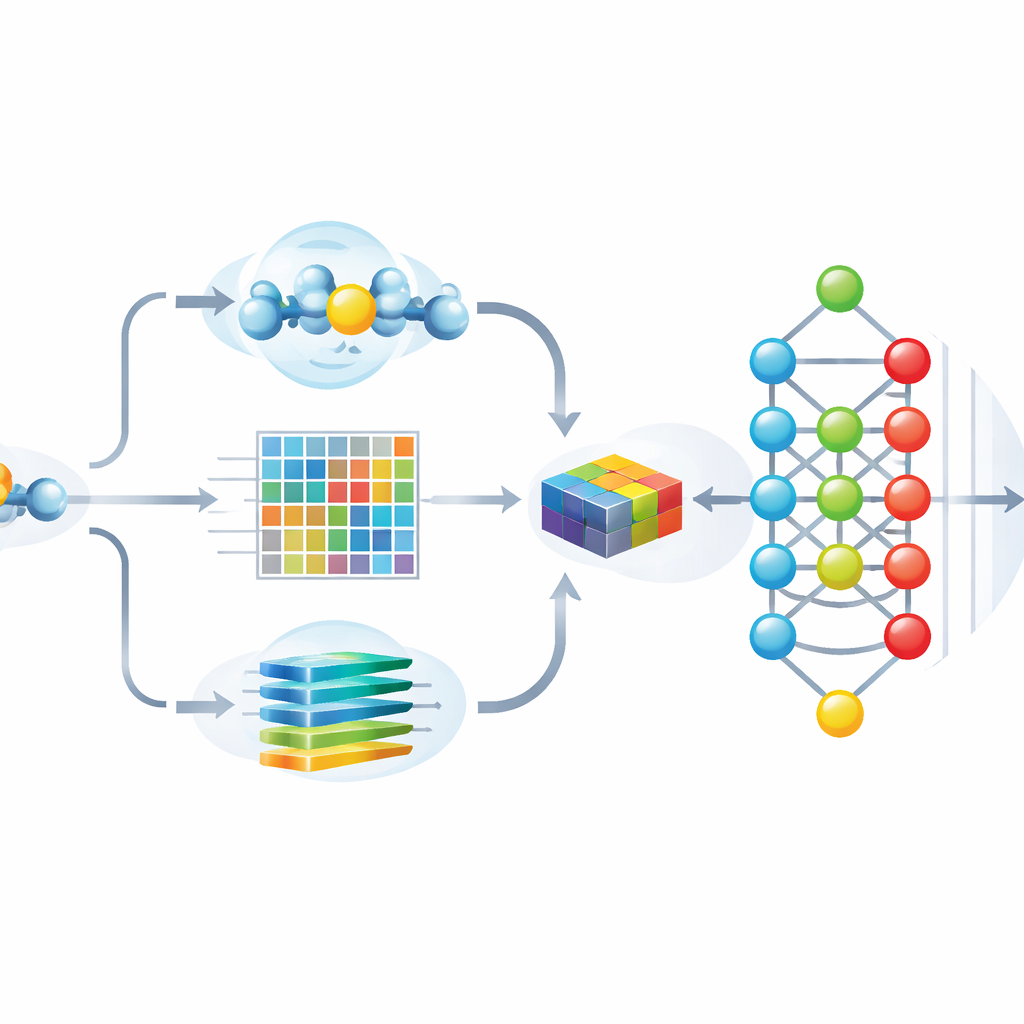
Making the model both accurate and explainable
Feeding more than a thousand features into a deep neural network can improve accuracy, but it also risks overfitting and makes it hard to see which clues the model truly relies on. To address this, the team uses a game-theory–inspired technique called SHAP to score how much each feature contributes to correct predictions. They then keep only the most informative 243 features, dramatically trimming redundancy while preserving performance. Visual analyses show that, after this selection, SUMO and non-SUMO sites form well-separated clusters, and the most important features align with intuitive properties such as surface exposure, local charge, and characteristic sequence patterns around the modified lysine.
Putting Hybrid-Sumo to the test
To guard against misleading results, the researchers carefully built balanced and imbalanced datasets from a curated protein modification database, removed near-duplicate sequences, and evaluated Hybrid-Sumo using repeated cross-validation as well as completely independent test sets. The final model reached about 99.7% accuracy on training data and around 96% accuracy on unseen proteins, slightly but consistently outperforming several strong deep learning and ensemble methods built specifically for the same task. Statistical tests confirmed that the gains from SHAP-based feature selection are real rather than due to chance, and comparisons with other common algorithms showed that the advantage comes from the hybrid features and careful optimization, not just from choosing a deep network.
What this means going forward
For non-specialists, the key message is that Hybrid-Sumo offers a more reliable way to predict where the SUMO tag will land on a protein, using a blend of 3D structure, evolutionary signals, and modern sequence “language” models. By reducing trial-and-error in the lab, it can help scientists prioritize experiments, explore how SUMOylation contributes to disease, and eventually guide therapies that target or exploit this subtle protein switch. The same design principles—combining diverse views of a molecule and then using interpretable feature selection—could also be adapted to forecast many other kinds of protein modifications that underlie health and disease.
Citation: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Keywords: SUMOylation, protein modification, deep learning, feature selection, bioinformatics