Clear Sky Science · fr
Cadre d’apprentissage profond avec sélection de caractéristiques interprétable pour une prédiction précise des sites de SUMOylation
Pourquoi cela compte pour la santé et la médecine
Les protéines pilotent presque tous les processus de nos cellules, et de petits marquages chimiques ajoutés après la synthèse d’une protéine peuvent en modifier profondément la fonction. L’un de ces marquages, appelé SUMO, a été lié au cancer, à la maladie d’Alzheimer et à d’autres pathologies graves. Identifier expérimentalement l’emplacement précis du SUMO sur des milliers de protéines est lent et coûteux. Cet article présente Hybrid-Sumo, un modèle informatique puissant capable de localiser avec grande précision les sites probables d’addition de SUMO, ce qui pourrait accélérer la recherche fondamentale et la découverte de médicaments.
Comment les cellules modulent le comportement des protéines
Nos cellules ajustent finement le comportement des protéines grâce à des modifications « post-production » appelées modifications post‑traductionnelles. La SUMOylation est l’une de ces modifications : une petite protéine modifieuse est fixée sur une lysine d’une protéine cible. Ce petit changement peut altérer le repliement de la protéine, sa localisation intracellulaire, sa durée de vie et ses interactions. Parce que la SUMOylation influence le contrôle des gènes, la réparation de l’ADN et le recyclage des protéines, elle joue un rôle central dans l’homéostasie cellulaire. Lorsqu’elle dysfonctionne, elle peut contribuer aux maladies neurodégénératives et au cancer, ce qui fait des cartographies précises des sites de SUMO une priorité pour les biologistes.
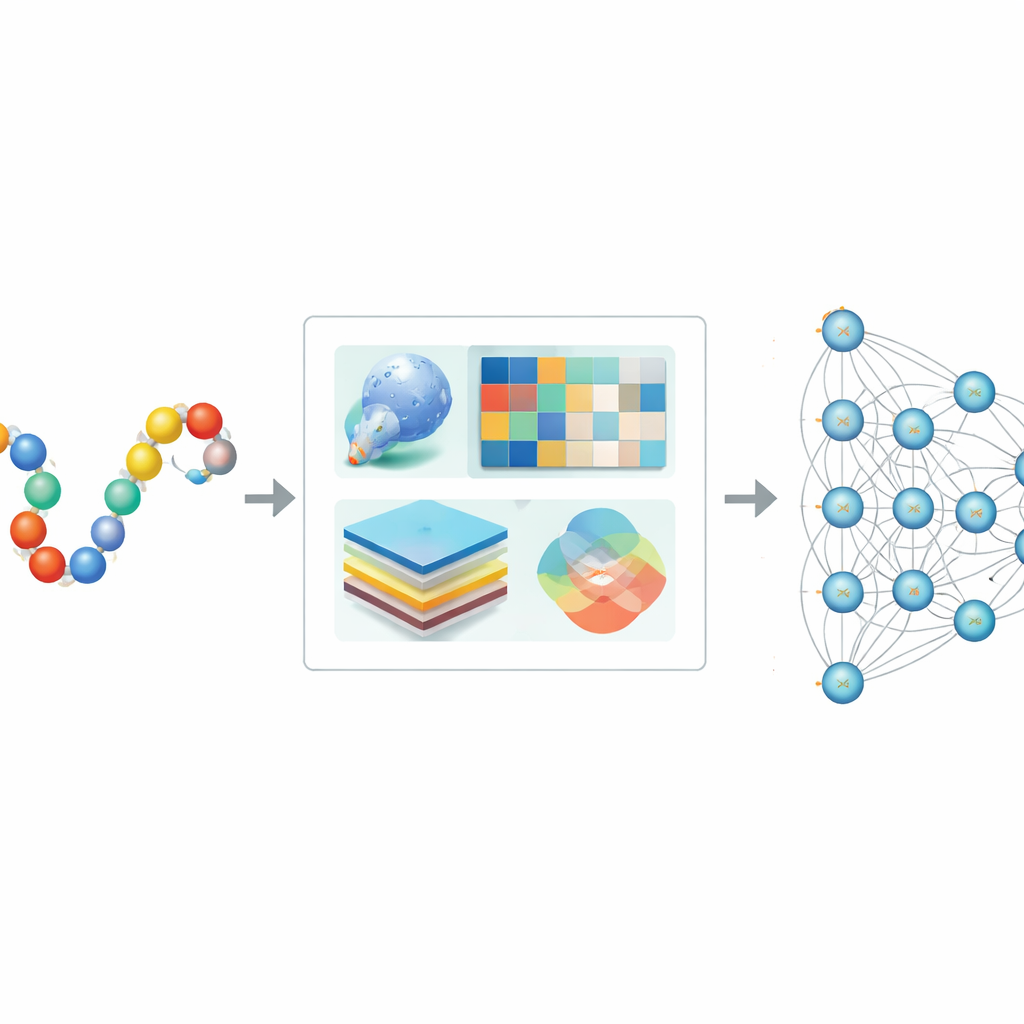
Pourquoi les ordinateurs sont nécessaires pour repérer les marquages SUMO
Les méthodes de laboratoire traditionnelles peuvent confirmer la SUMOylation, mais elles ne sont pas adaptées à l’échelle du grand nombre de protéines chez l’humain et d’autres organismes. Les outils informatiques antérieurs tentaient de repérer les sites SUMO en reconnaissant de courts motifs de séquence ou en utilisant des méthodes classiques d’apprentissage automatique, comme les arbres de décision ou les machines à vecteurs de support. Bien qu’utiles, ces approches négligeaient souvent le contexte complet d’une protéine, incluant sa structure tridimensionnelle et son histoire évolutive, et elles peinaient lorsque les données étaient déséquilibrées, avec beaucoup moins de sites SUMO connus que de sites non-SUMO. En conséquence, les prédictions pouvaient être biaisées ou incapables de généraliser à de nouvelles protéines.
Une vue hybride de chaque site protéique
Les auteurs ont conçu Hybrid-Sumo pour examiner chaque site potentiel de SUMO selon trois angles complémentaires. D’abord, ils utilisent une mesure structurelle appelée exposition demi-sphère pour estimer dans quelle mesure un résidu est enfoui ou exposé à la surface de la protéine, ce qui influence l’accès physique du SUMO. Ensuite, ils calculent des profils évolutifs qui capturent l’évolution d’une position parmi des protéines apparentées, puis compressent ces profils avec des méthodes par ondelettes pour mettre en évidence les motifs importants tout en réduisant le bruit. Enfin, ils exploitent un modèle Transformer initialement développé pour le langage, traitant les séquences d’acides aminés comme des phrases afin que le réseau apprenne des « embeddings » contextuels riches décrivant la relation de chaque position avec ses voisines le long de la chaîne. Ces trois ensembles de valeurs sont fusionnés en une description détaillée de chaque site.
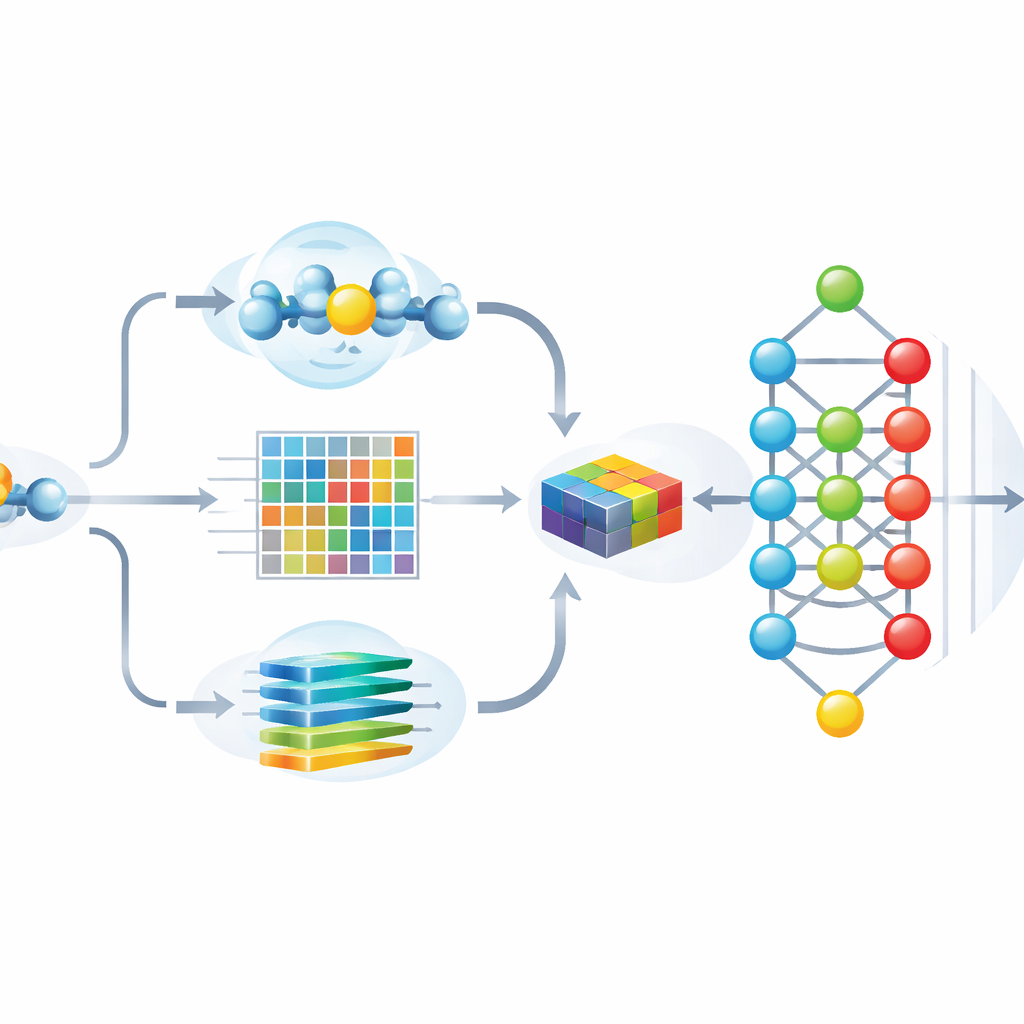
Rendre le modèle à la fois précis et explicable
Alimenter un réseau de neurones profond avec plus d’un millier de caractéristiques peut améliorer la précision, mais augmente aussi le risque de surapprentissage et rend difficile l’identification des indices réellement utilisés par le modèle. Pour y remédier, l’équipe utilise une technique inspirée de la théorie des jeux appelée SHAP pour évaluer la contribution de chaque caractéristique aux bonnes prédictions. Ils conservent ensuite seulement les 243 caractéristiques les plus informatives, réduisant fortement la redondance tout en préservant les performances. Des analyses visuelles montrent qu’après cette sélection, les sites SUMO et non-SUMO forment des groupes bien séparés, et que les caractéristiques les plus importantes correspondent à des propriétés intuitives telles que l’exposition à la surface, la charge locale et les motifs de séquence caractéristiques autour de la lysine modifiée.
Évaluer Hybrid-Sumo
Pour éviter des résultats trompeurs, les chercheurs ont construit avec soin des jeux de données équilibrés et déséquilibrés à partir d’une base de données annotée de modifications protéiques, éliminé les séquences quasi dupliquées, et évalué Hybrid-Sumo via une validation croisée répétée ainsi que des jeux de tests complètement indépendants. Le modèle final a atteint environ 99,7 % de précision sur les données d’entraînement et environ 96 % sur des protéines non vues auparavant, dépassant légèrement mais de manière constante plusieurs méthodes solides d’apprentissage profond et d’ensemble conçues pour la même tâche. Des tests statistiques ont confirmé que les gains apportés par la sélection de caractéristiques basée sur SHAP sont réels et non dus au hasard, et des comparaisons avec d’autres algorithmes courants ont montré que l’avantage provient des caractéristiques hybrides et de l’optimisation soignée, et non seulement du choix d’un réseau profond.
Ce que cela implique pour l’avenir
Pour un public non spécialiste, le message principal est que Hybrid-Sumo offre un moyen plus fiable de prédire où le marquage SUMO s’attachera sur une protéine, en combinant structure 3D, signaux évolutifs et modèles « linguistiques » modernes pour les séquences. En réduisant les essais-erreurs au laboratoire, il peut aider les scientifiques à prioriser les expériences, explorer le rôle de la SUMOylation dans les maladies, et à terme orienter des thérapies ciblant ou exploitant cet interrupteur protéique subtil. Les mêmes principes de conception — combiner des vues diverses d’une molécule puis appliquer une sélection de caractéristiques interprétable — pourraient également être adaptés pour prévoir de nombreuses autres modifications protéiques impliquées dans la santé et la maladie.
Citation: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Mots-clés: SUMOylation, modification des protéines, apprentissage profond, sélection de caractéristiques, bioinformatique