Clear Sky Science · pt
Estrutura de aprendizado profundo com seleção de características interpretável para previsão precisa de sítios de SUMOilação
Por que isso importa para a saúde e a medicina
Proteínas controlam quase todos os processos em nossas células, e pequenos rótulos químicos adicionados após a síntese de uma proteína podem alterar completamente sua função. Um desses rótulos, chamado SUMO, foi associado ao câncer, à doença de Alzheimer e a outras condições graves. Identificar experimentalmente exatamente onde o SUMO se liga em milhares de proteínas é um processo lento e caro. Este artigo apresenta o Hybrid-Sumo, um modelo computacional poderoso que pode localizar com notável precisão os prováveis sítios de ligação do SUMO, potencialmente acelerando a pesquisa básica e a descoberta de fármacos no futuro.
Como as células mudam o comportamento das proteínas
Nossas células ajustam finamente o comportamento das proteínas usando edições “pós-produção” conhecidas como modificações pós-traducionais. A SUMOilação é uma dessas edições em que uma pequena proteína modificadora é anexada a um resíduo de lisina na proteína alvo. Essa pequena alteração pode modificar como a proteína se dobra, para onde ela se desloca na célula, quanto tempo ela persiste e com quais parceiros ela se liga. Como a SUMOilação influencia o controle gênico, a reparação do DNA e a reciclagem de proteínas, ela está profundamente envolvida na manutenção do equilíbrio celular. Quando a SUMOilação falha, pode contribuir para doenças neurodegenerativas e câncer, tornando mapas precisos dos sítios de SUMO uma prioridade alta para os biólogos.
Por que computadores são necessários para encontrar os rótulos SUMO
Métodos de laboratório tradicionais podem confirmar a SUMOilação, mas não escalam bem para o grande número de proteínas em humanos e outros organismos. Ferramentas computacionais anteriores tentaram detectar sítios de SUMO reconhecendo padrões curtos de sequência ou usando métodos clássicos de aprendizado de máquina, como árvores de decisão ou máquinas de vetor de suporte. Embora úteis, essas abordagens muitas vezes ignoraram o contexto completo de uma proteína, incluindo sua forma tridimensional e história evolutiva, e tiveram dificuldade quando os dados eram desbalanceados, com muito menos sítios SUMO conhecidos do que sítios não-SUMO. Como resultado, as previsões podiam ficar enviesadas ou falhar ao generalizar para novas proteínas.
Uma visão híbrida de cada sítio proteico
Os autores projetaram o Hybrid-Sumo para observar cada sítio potencial de SUMO a partir de três ângulos complementares ao mesmo tempo. Primeiro, eles usam uma medida estrutural chamada exposição em meia-esfera (half-sphere exposure) para estimar o quão enterrado ou exposto um resíduo está na superfície da proteína, o que afeta se o SUMO pode alcançá-lo fisicamente. Segundo, computam perfis evolutivos que capturam como uma posição mudou entre proteínas relacionadas e então comprimem esses perfis usando métodos wavelet para destacar padrões importantes reduzindo o ruído. Terceiro, aproveitam um modelo transformer originalmente construído para linguagem, tratando sequências de aminoácidos como frases para que a rede aprenda ricos “embeddings” contextuais que descrevem como cada posição se relaciona com seus vizinhos ao longo da cadeia. Esses três conjuntos de valores são fundidos em uma única descrição detalhada de cada sítio.
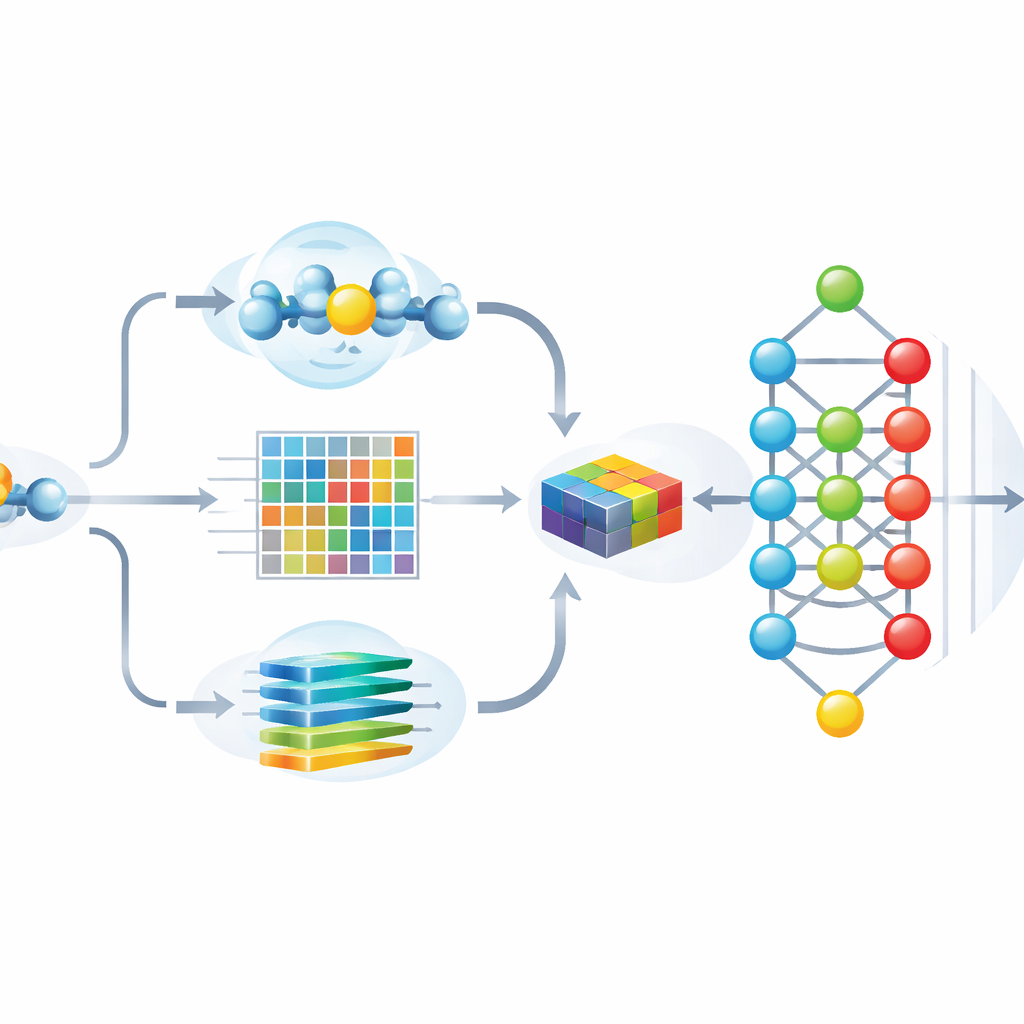
Tornando o modelo ao mesmo tempo preciso e explicável
Fornecer mais de mil características a uma rede neural profunda pode melhorar a precisão, mas também aumenta o risco de overfitting e dificulta ver em quais pistas o modelo realmente se baseia. Para resolver isso, a equipe usa uma técnica inspirada na teoria dos jogos chamada SHAP para pontuar quanto cada característica contribui para previsões corretas. Eles então mantêm apenas as 243 características mais informativas, reduzindo dramaticamente redundâncias enquanto preservam o desempenho. Análises visuais mostram que, após essa seleção, sítios SUMO e não-SUMO formam agrupamentos bem separados, e as características mais importantes alinham-se com propriedades intuitivas, como exposição superficial, carga local e padrões de sequência característicos ao redor da lisina modificada.
Testando o Hybrid-Sumo
Para se proteger contra resultados enganadores, os pesquisadores construíram cuidadosamente conjuntos de dados balanceados e desbalanceados a partir de um banco de dados curado de modificações proteicas, removeram sequências quase duplicadas e avaliaram o Hybrid-Sumo usando validação cruzada repetida, bem como conjuntos de teste completamente independentes. O modelo final alcançou cerca de 99,7% de acurácia nos dados de treinamento e aproximadamente 96% em proteínas não vistas, superando de forma consistente, ainda que por pouco, vários métodos fortes de aprendizado profundo e ensemble projetados especificamente para a mesma tarefa. Testes estatísticos confirmaram que os ganhos obtidos com a seleção de características baseada em SHAP são reais e não fruto do acaso, e comparações com outros algoritmos comuns mostraram que a vantagem vem das características híbridas e da otimização cuidadosa, não apenas da escolha de uma rede profunda.
O que isso significa daqui para frente
Para não especialistas, a mensagem principal é que o Hybrid-Sumo oferece uma maneira mais confiável de prever onde o rótulo SUMO se fixará em uma proteína, usando uma combinação de estrutura 3D, sinais evolutivos e modelos modernos de “linguagem” de sequência. Ao reduzir tentativa e erro no laboratório, ele pode ajudar cientistas a priorizar experimentos, explorar como a SUMOilação contribui para doenças e, eventualmente, orientar terapias que visem ou aproveitem esse interruptor sutil da proteína. Os mesmos princípios de projeto — combinar visões diversas de uma molécula e então usar seleção de características interpretável — também podem ser adaptados para prever muitos outros tipos de modificações proteicas que sustentam a saúde e a doença.
Citação: Alyahya, A.N., Khan, S., Dilshad, N. et al. Deep learning framework with interpretable feature selection for accurate SUMOylation site prediction. Sci Rep 16, 10419 (2026). https://doi.org/10.1038/s41598-026-41489-0
Palavras-chave: SUMOilação, modificação de proteína, aprendizado profundo, seleção de características, bioinformática