Clear Sky Science · sv
Upptäckt av kända genfusioner i cancercellinjer med hjälp av helgenom-bisulfitsekvenseringsdata
Varför ett test som gör mer spelar roll
Onkologer vill allt oftare utvinna flera typer av information från ett enda DNA-prov: vilka gener som är aktiva eller tysta, hur DNA:t är veckat eller kopierat, och om viktiga gener har kapats och satts ihop på skadliga sätt. I dag kräver dessa frågor vanligtvis flera olika laboratorietester, som var och en förbrukar värdefullt material och ökar kostnaden. Denna studie undersöker om ett allmänt använt test för kemiska markörer på DNA, så kallad helgenom-bisulfitsekvensering, också kan avslöja genfusioner — onormala sammanfogningar av två gener som ofta driver cancer och styr behandlingsval.
Underliga genkombinationer i cancer
Genfusioner uppstår när kromosomer bryts och fästs om på fel platser, vilket syr ihop delar av två olika gener. Dessa kombinationer kan överladda tillväxtsignaler, tysta skyddande gener eller skapa hybrida proteiner som driver celler mot cancer. Kända exempel är BCR–ABL1-fusionen vid kronisk myeloisk leukemi och fusioner i prostatacancer och lungcancer som avgör vilka läkemedel som fungerar. På grund av deras kliniska betydelse letar laboratorier vanligtvis efter fusioner med tester riktade mot RNA eller genom att skanna hela genomet, inte med metoder avsedda för att läsa kemiska märken på DNA.
En kraftfull men underutnyttjad datakälla
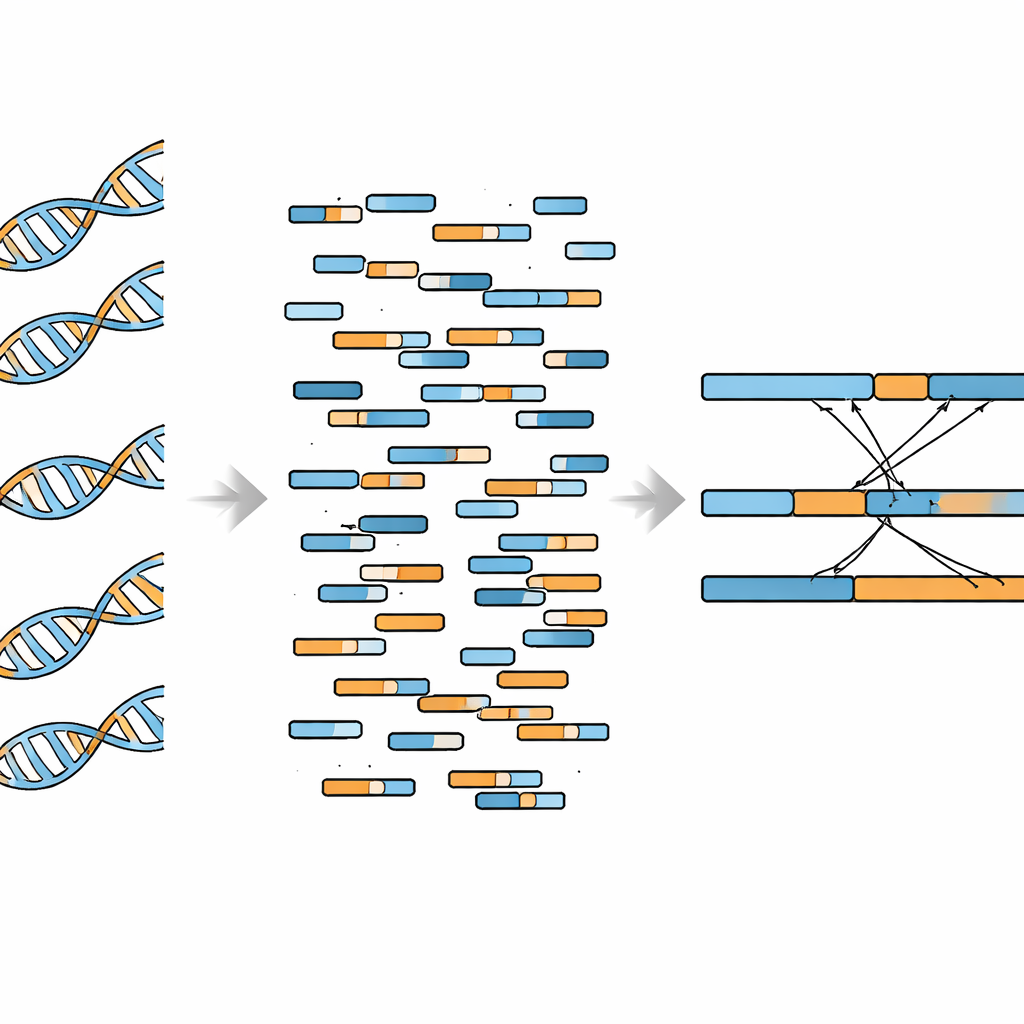
Helgenom-bisulfitsekvensering (WGBS) skapades för att kartlägga DNA-metylering — små kemiska märken som hjälper till att styra vilka gener som är aktiva. Den har blivit ett arbetsredskap både inom grundforskning och i likvortester som analyserar DNA-fragment i blodomloppet. Från en enda WGBS-körning kan forskare redan utvinna information om metylering, kopieringsnummerförändringar, enkelbasmutationer och hur DNA delas upp i fragment. Däremot har fusiondetektion saknats från den listan. Hindret är att den kemiska behandlingen som används i WGBS bryter DNA:t till kortare bitar och omvandlar många cytosinbaser, vilket gör de resulterande sekvenserna svårare att få att linjera exakt mot referensgenomet.
Lära fusion-sökningsverktyg en ny finess
Författarna satte som mål att anpassa WGBS-data för fusionupptäckt genom att använda programvara som förstår bisulfitbehandlat DNA:s egenheter och kan hantera "split"-läsningar — korta sekvenser som kartlägger delvis till en gen och delvis till en annan. De fokuserade på kända fusionhändelser istället för att söka blint över hela genomet. Först testade de sin pipeline på K562-leukemiceller, som bär på den välstuderade BCR–ABL1-fusionen. Brytpunkter bestämda från WGBS matchade väl dem från traditionell helgenomsekvensering, och täckningsdippar runt fusionsstället syntes i båda datatyperna. Forskarna visade också att WGBS kunde upptäcka fusionen pålitligt när så lite som cirka 8 % av DNA:t i ett prov kom från fusionpositiva celler, med perfekt detektion när den andelen nådde 10 % vid relativt djup sekvensering.
Skalning upp till många fusioner samtidigt
Nästa fråga var om deras metod kunde hantera flera fusioner i samma prov. De tillämpade den på MCF-7 bröstcancerceller, som rymmer ett dussin tidigare bekräftade genfusioner. Deras WGBS-baserade metod återfann 10 av dessa 12 fusioner, inklusive omarrangemang inom en enskild kromosom och sådana som bryggar mellan två olika kromosomer. Antalet stödjande läsningar var mycket konsekvent över upprepade experiment, vilket visar att metoden är tekniskt stabil. Samtidigt identifierade författarna en viktig begränsning: när de tittade utanför de specifika genregionerna var bakgrundssignaler som liknade falska fusioner mycket vanligare än verkliga. Det tyder på att WGBS fungerar bäst för riktade kontroller av kända fusioner snarare än för öppna, sökande insatser.
Löften och praktiska begränsningar
Även om resultaten är uppmuntrande belyser de också begränsningar. Tester gjordes i homogena cellinjer, inte i verkliga patientprover där cancerceller är mer blandade och fusionbrytpunkter kan skilja sig mellan individer. Upptäcktsgränsen på strax över 8 % tumör-DNA passar väl för vävnadsbiopsier, där cancerceller ofta dominerar, men kan vara otillräcklig för blodbaserade tester som måste hitta mycket små mängder tumör-DNA. Att uppnå nödvändig sekvenseringsdjup är också kostsamt, och vissa fusioner som faller utanför kända gengränser kommer fortfarande att missas av designen.
Vad detta betyder för framtida cancertester
Denna studie visar att en datamängd som ursprungligen samlades in för att läsa kemiska märken på DNA kan användas om för att upptäcka viktiga genfusioner, åtminstone när dessa fusioner är kända i förväg och finns i rimliga nivåer. För forskare och kliniker som redan använder WGBS innebär detta att mer värde kan utvinnas ur varje experiment: metyleringsmönster, andra genetiska förändringar och nu även fusionshändelser kan alla bedömas från samma körning. Med ytterligare förbättringar, validering i patientprover och smartare mjukvara kan WGBS bli ett centralt test som levererar flera lager av insikt om hur en cancer är uppbyggd — och hur den bäst behandlas — från ett enda värdefullt prov.
Citering: Kim, T., Bang, D. Detection of known gene fusions in cancer cell lines using whole-genome bisulfite sequencing data. Sci Rep 16, 13254 (2026). https://doi.org/10.1038/s41598-026-40803-0
Nyckelord: genfusion, DNA-metylering, helgenom-bisulfitsekvensering, cancergenomik, likvortest (liquid biopsy)