Clear Sky Science · en
Detection of known gene fusions in cancer cell lines using whole-genome bisulfite sequencing data
Why one test that does more matters
Cancer doctors increasingly want to learn many things from a single sample of DNA: which genes are switched on or off, how the DNA is folded or copied, and whether key genes have been cut and pasted together in harmful ways. Today, those questions usually require several different laboratory tests, each using up precious material and adding cost. This study asks whether one widely used test for chemical tags on DNA, called whole-genome bisulfite sequencing, can also reveal gene fusions — the abnormal joinings of two genes that often drive cancer and guide treatment choices.
Strange gene mash-ups in cancer
Gene fusions arise when chromosomes break and reattach in the wrong places, splicing together pieces of two different genes. These mash-ups can supercharge growth signals, silence protective genes, or create hybrid proteins that push cells toward cancer. Famous examples include the BCR–ABL1 fusion in chronic myeloid leukemia and fusions in prostate and lung cancers that determine which drugs will work. Because of their clinical importance, laboratories typically hunt for fusions using tests aimed specifically at RNA or by scanning the entire genome, not with methods built for reading chemical marks on DNA.
A powerful but underused data source
Whole-genome bisulfite sequencing (WGBS) was designed to map DNA “methylation” — small chemical tags that help control which genes are active. It has become a workhorse for both basic research and liquid biopsy tests that analyze fragments of DNA in the bloodstream. From one WGBS run, scientists can already extract information on methylation, copy number changes, single-letter mutations, and how DNA is chopped into fragments. However, fusion detection has been missing from that list. The obstacle is that the chemical treatment used in WGBS breaks DNA into shorter pieces and converts many cytosine bases, making the resulting sequences harder to line up accurately on the reference genome.
Teaching fusion-hunting tools a new trick
The authors set out to retrofit WGBS data for fusion discovery by using software that understands the quirks of bisulfite-treated DNA and can handle “split” reads — short sequences that map partly to one gene and partly to another. They focused on known fusion events rather than searching blindly across the entire genome. First, they tested their pipeline on K562 leukemia cells, which carry the well-studied BCR–ABL1 fusion. Breakpoints pinpointed from WGBS closely matched those from traditional whole-genome sequencing, and coverage dips around the fusion site appeared in both data types. The team also showed that WGBS could detect the fusion reliably when as little as about 8% of the DNA in a sample came from fusion-positive cells, with perfect detection once this fraction reached 10% at relatively deep sequencing.
Scaling up to many fusions at once
Next, the researchers asked whether their approach could handle several fusions in the same sample. They applied it to MCF-7 breast cancer cells, which harbor a dozen previously confirmed gene fusions. Their WGBS-based method recovered 10 of these 12 fusions, spanning both rearrangements within a single chromosome and those bridging two different chromosomes. Counts of supporting reads were highly consistent across repeated experiments, showing that the method is technically stable. At the same time, the authors uncovered a key limitation: when they looked outside the specific gene regions of interest, background signals resembling false fusions were much more common than true ones. This suggests that WGBS works best for targeted checks of known fusions rather than for open-ended fishing expeditions.
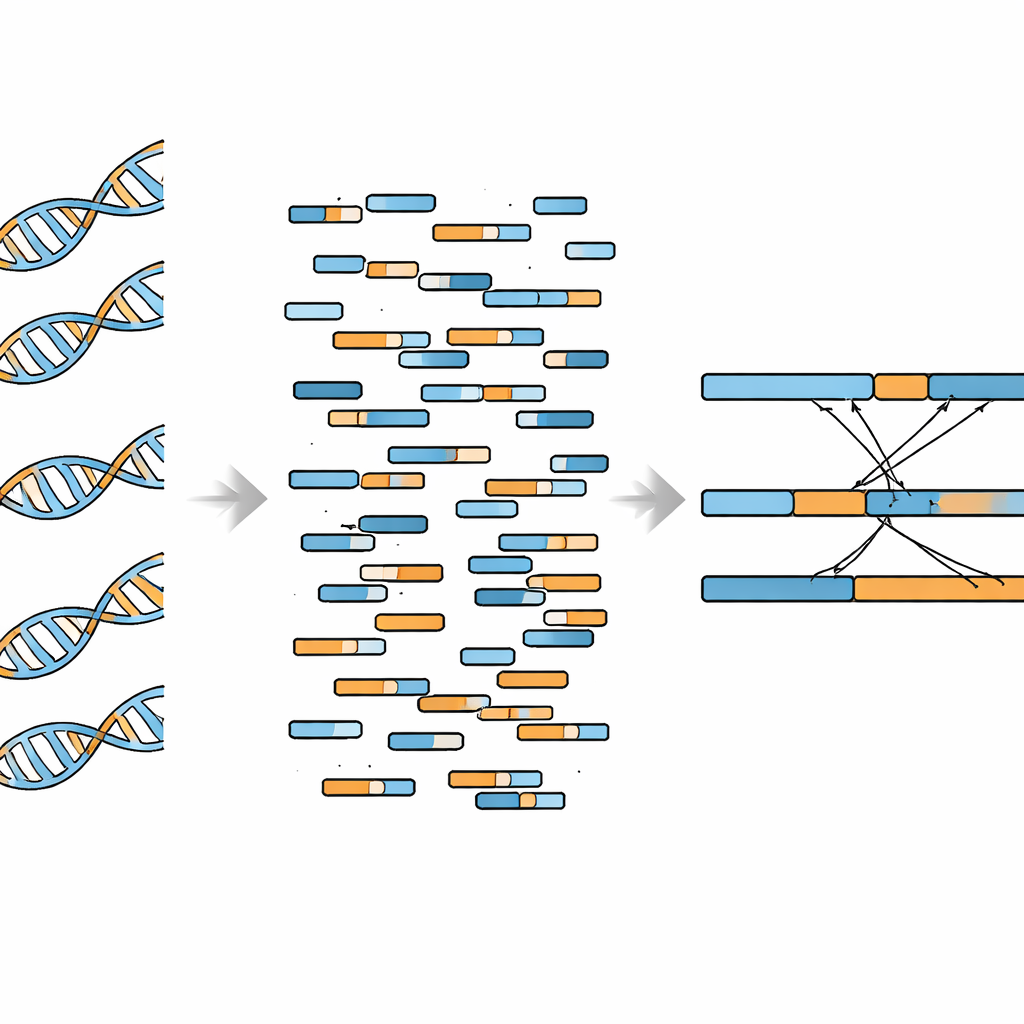
Promise and practical limits
While encouraging, the results also highlight boundaries. The tests were performed in uniform cell lines, not in real patient samples where cancer cells are more mixed and fusion breakpoints can vary from person to person. The detection threshold of just over 8% tumor-derived DNA is well suited to tissue biopsies, where cancer cells often dominate, but may fall short for blood-based tests that must sift through trace amounts of tumor DNA. Achieving the required sequencing depth is also expensive, and some fusions that fall outside known gene boundaries will still be missed by design.
What this means for future cancer testing
This study shows that a data set originally collected to read chemical tags on DNA can be repurposed to spot important gene fusions, at least when those fusions are known in advance and present at reasonable levels. For researchers and clinicians already using WGBS, this means more value can be extracted from each experiment: methylation patterns, other genetic changes, and now fusion events can all be assessed from the same run. With further refinement, validation in patient samples, and smarter software, WGBS could become a central hub assay that feeds many layers of insight into how a cancer is built — and how best to treat it — from one precious sample.
Citation: Kim, T., Bang, D. Detection of known gene fusions in cancer cell lines using whole-genome bisulfite sequencing data. Sci Rep 16, 13254 (2026). https://doi.org/10.1038/s41598-026-40803-0
Keywords: gene fusion, DNA methylation, whole-genome bisulfite sequencing, cancer genomics, liquid biopsy