Clear Sky Science · pl
Wykrywanie znanych fuzyjnych genów w liniach komórkowych nowotworów przy użyciu danych z całogenomowego sekwencjonowania bisulfitem
Dlaczego ważne jest jedno badanie dające więcej informacji
Onkolodzy coraz częściej chcą wyciągnąć z jednej próbki DNA jak najwięcej informacji: które geny są włączone lub wyłączone, jak DNA jest złożony lub skopiowany oraz czy kluczowe geny zostały nieprawidłowo poskładane razem. Dziś odpowiedzi na te pytania zwykle wymagają kilku różnych badań laboratoryjnych, z których każde zużywa cenny materiał i zwiększa koszty. W tym badaniu sprawdzono, czy jedno powszechnie stosowane badanie chemicznych znaczników na DNA, zwane całogenomowym sekwencjonowaniem bisulfitem, może także ujawnić fuzje genów — nieprawidłowe łączenia dwóch genów, które często napędzają rozwój nowotworu i wpływają na wybór terapii.
Dziwne zlepki genów w nowotworach
Fuzje genów powstają, gdy chromosomy pękają i ponownie łączą się w niewłaściwych miejscach, składjąc fragmenty dwóch różnych genów. Takie zlepki mogą nadmiernie wzmacniać sygnały wzrostu, wyciszać geny ochronne lub tworzyć hybrydowe białka, które skłaniają komórki do transformacji nowotworowej. Słynne przykłady to fuzja BCR–ABL1 w przewlekłej białaczce szpikowej oraz fuzje w raku prostaty i płuca, które decydują, które leki będą skuteczne. Ze względu na ich znaczenie kliniczne laboratoria zazwyczaj poszukują fuzji za pomocą testów ukierunkowanych na RNA lub przez skanowanie całego genomu, a nie metod zaprojektowanych do odczytu chemicznych znaków na DNA.
Mocne, lecz niedocenione źródło danych
Całogenomowe sekwencjonowanie bisulfitem (WGBS) zostało opracowane do mapowania „metylacji” DNA — drobnych znaczników chemicznych, które pomagają kontrolować aktywność genów. Stało się narzędziem pracy zarówno w badaniach podstawowych, jak i w testach płynnej biopsji analizujących fragmenty DNA we krwi. Z jednego przebiegu WGBS naukowcy potrafią już wyciągnąć informacje o metylacji, zmianach liczby kopii, pojedynczych mutacjach oraz o tym, jak DNA jest cięte na fragmenty. Jednak wykrywanie fuzji brakowało na tej liście. Przeszkodą jest to, że zabieg chemiczny stosowany w WGBS łamie DNA na krótsze fragmenty i przekształca wiele zasad cytozynowych, co utrudnia dokładne dopasowanie otrzymanych sekwencji do genomu referencyjnego.
Nauczanie narzędzi detekcji fuzji nowej sztuczki
Autorzy postanowili dostosować dane WGBS do wykrywania fuzji, używając oprogramowania, które rozumie specyfikę DNA poddanego bisulfitem i potrafi obsługiwać „split” ready — krótkie odczyty mapujące się częściowo do jednego genu, a częściowo do innego. Skupili się na znanych zdarzeniach fuzyjnych zamiast na bezcelowym przeszukiwaniu całego genomu. Najpierw przetestowali swój pipeline na komórkach białaczkowych K562, które niosą dobrze poznaną fuzję BCR–ABL1. Punkty przerwań wyznaczone z WGBS były bliskie tym z tradycyjnego sekwencjonowania całogenomowego, a spadki pokrycia wokół miejsca fuzji pojawiały się w obu typach danych. Zespół wykazał także, że WGBS potrafi wykryć fuzję niezawodnie, gdy zaledwie około 8% DNA w próbce pochodzi z komórek z fuzją, przy czym wykrycie było perfekcyjne, gdy ten udział osiągnął 10% przy stosunkowo głębokim sekwencjonowaniu.
Skalowanie do wielu fuzji jednocześnie
Następnie badacze sprawdzili, czy ich podejście poradzi sobie z kilkoma fuzjami w tej samej próbce. Zastosowali je w komórkach raka piersi MCF-7, które zawierają kilkanaście wcześniej potwierdzonych fuzji genów. Metoda oparta na WGBS odzyskała 10 z tych 12 fuzji, obejmując zarówno przestawienia wewnątrz jednego chromosomu, jak i łączenia pomiędzy dwoma różnymi chromosomami. Liczby wspierających odczytów były wysokospójne w powtarzanych eksperymentach, co pokazuje, że metoda jest technicznie stabilna. Jednocześnie autorzy ujawnili istotne ograniczenie: poza specyficznymi regionami genowymi zainteresowania sygnały tła przypominające fałszywe fuzje były znacznie częstsze niż prawdziwe. Sugeruje to, że WGBS najlepiej nadaje się do ukierunkowanej weryfikacji znanych fuzji, a nie do otwartego przeszukiwania.
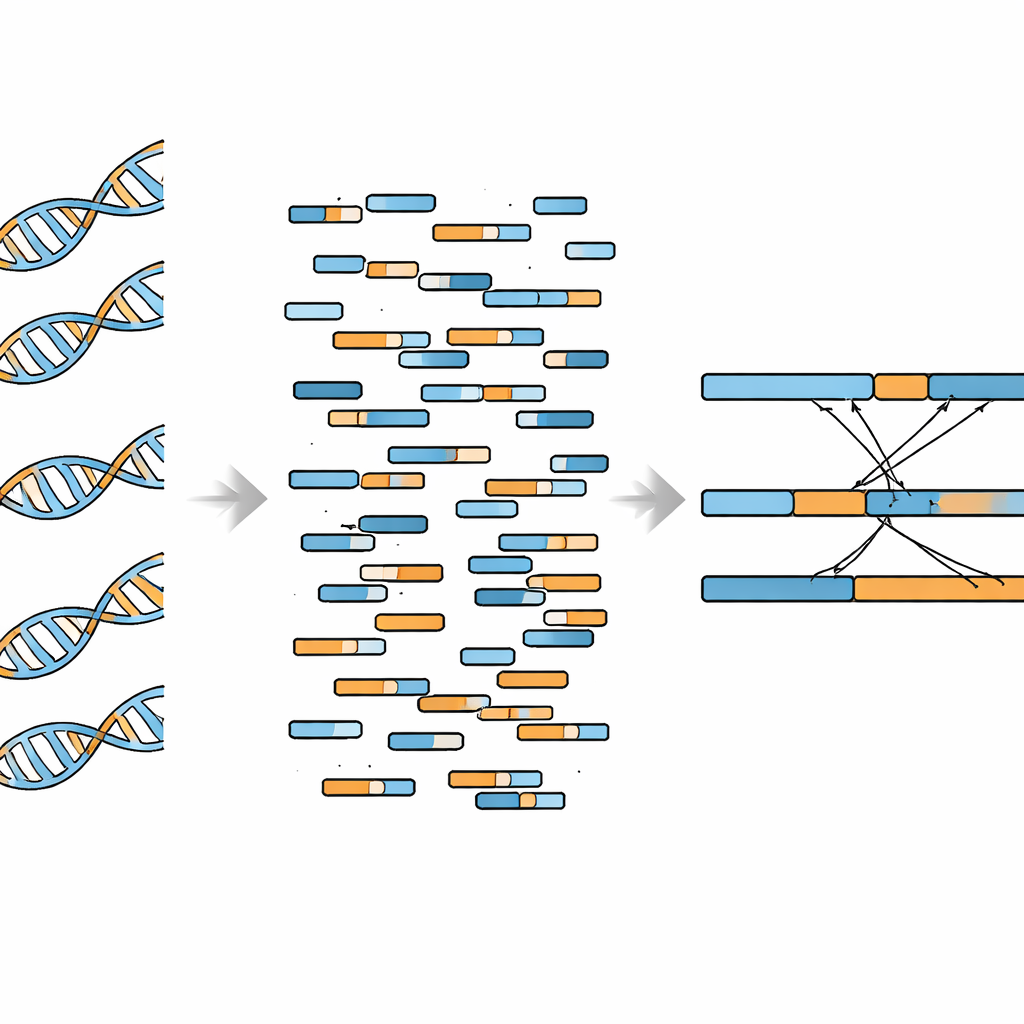
Obietnica i praktyczne ograniczenia
Choć wyniki są zachęcające, podkreślają też ograniczenia. Testy przeprowadzono na jednorodnych liniach komórkowych, a nie na prawdziwych próbkach pacjentów, gdzie komórki nowotworowe są bardziej wymieszane, a miejsca przerwań fuzji mogą się różnić między osobami. Próg detekcji nieco ponad 8% DNA pochodzącego z guza dobrze pasuje do biopsji tkankowych, w których komórki nowotworowe często dominują, ale może być niewystarczający dla testów z krwi, które muszą wyłuskać śladowe ilości DNA nowotworowego. Osiągnięcie wymaganej głębokości sekwencjonowania jest też kosztowne, a niektóre fuzje wychodzące poza znane granice genów nadal będą pomijane przez konstrukcję metody.
Co to oznacza dla przyszłych badań diagnostycznych w onkologii
Badanie pokazuje, że zestaw danych pierwotnie zebrany do odczytu chemicznych znaczników na DNA można ponownie wykorzystać do wykrywania ważnych fuzji genów, przynajmniej gdy te fuzje są znane z wyprzedzeniem i obecne na wystarczającym poziomie. Dla badaczy i klinicystów już korzystających z WGBS oznacza to większą wartość z każdej próby: wzorce metylacji, inne zmiany genetyczne i teraz zdarzenia fuzyjne można ocenić z tego samego przebiegu. Przy dalszym dopracowaniu, walidacji na próbkach pacjentów i inteligentniejszym oprogramowaniu WGBS mógłby stać się centralnym badaniem dostarczającym wiele warstw informacji o budowie nowotworu — i o tym, jak go najlepiej leczyć — z jednej cennej próbki.
Cytowanie: Kim, T., Bang, D. Detection of known gene fusions in cancer cell lines using whole-genome bisulfite sequencing data. Sci Rep 16, 13254 (2026). https://doi.org/10.1038/s41598-026-40803-0
Słowa kluczowe: fuzja genów, metylacja DNA, całogenomowe sekwencjonowanie bisulfitem, genomika nowotworów, płynna biopsja