Clear Sky Science · ja
全ゲノムビスルフィトシーケンシングデータを用いたがん細胞株における既知の遺伝子融合の検出
1つの検査で多くを調べられる意義
がん診療では、1つのDNAサンプルから複数の情報を得たいという要求が高まっています:どの遺伝子がオン/オフになっているか、DNAがどう折りたたまれ複製されているか、重要な遺伝子が有害な形で切断・結合されていないか、などです。現在はこれらの質問に通常、複数の異なる検査が必要であり、貴重な試料を消費しコストも増えます。本研究は、DNAの化学的タグを調べるために広く使われている検査である全ゲノムビスルフィトシーケンシング(WGBS)が、がんを駆動し治療選択に影響することが多い遺伝子融合を明らかにできるかを検証しています。
がんに現れる奇妙な遺伝子の寄せ集め
遺伝子融合は、染色体が折れて誤った場所で再結合することで生じ、2つの異なる遺伝子の断片がつながります。これらの融合により増殖シグナルが強化されたり、保護的な遺伝子が沈黙したり、細胞をがんへと押しやるハイブリッドタンパク質が生まれることがあります。有名な例としては慢性骨髄性白血病のBCR–ABL1融合や、前立腺癌や肺癌に見られる薬剤選択を左右する融合があります。その臨床的重要性から、検査室では通常、RNAに焦点を当てた検査や全ゲノムをスキャンする方法で融合を探索し、DNAの化学的マークを読むために設計された手法では行われていません。
強力だが十分に活用されていないデータ源
全ゲノムビスルフィトシーケンシング(WGBS)は、どの遺伝子が活性化されているかを制御する小さな化学タグであるDNA「メチル化」をマップするために設計されました。基礎研究や血流中のDNA断片を解析するリキッドバイオプシー検査の主力技術になっています。1回のWGBS解析からは、メチル化、コピー数変化、一塩基変異、DNAの断片化パターンといった情報をすでに抽出できます。しかし、融合検出はそのリストから欠けていました。障害となっているのは、WGBSで用いる化学処理がDNAを短い断片に分解し多くのシトシン塩基を変換してしまうため、得られた配列を参照ゲノムに正確に整列させるのが難しくなる点です。
融合探索ツールに新たな技を教える
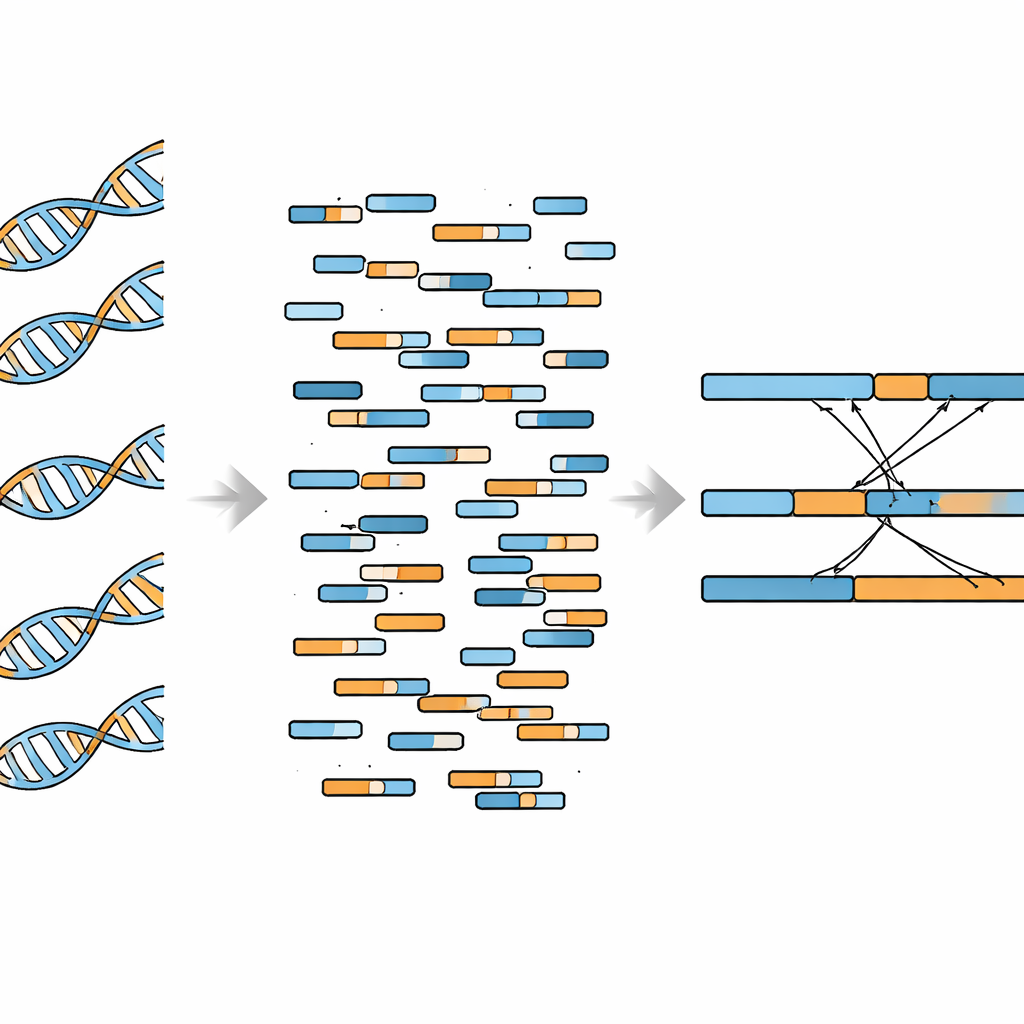
著者らは、ビスルフィト処理されたDNAの特性を理解し、「スプリット」リード(配列の一部がある遺伝子に、別の一部が別の遺伝子にマップされる短い配列)を扱えるソフトウェアを用いて、WGBSデータを融合検出に適合させることを目指しました。彼らは全ゲノムを無差別に探索するのではなく、既知の融合事象に注目しました。まず、よく研究されているBCR–ABL1融合を持つK562白血病細胞でパイプラインを検証しました。WGBSから特定された切断点は従来の全ゲノムシーケンスとよく一致し、融合部位付近のカバレッジ低下も両データで確認されました。さらに、WGBSはサンプル中の約8%程度が融合陽性細胞由来であれば融合を検出でき、比較的深いシーケンシングを行うとその割合が10%に達した時点で完全検出が得られることを示しました。
同時に多数の融合へ拡張する
次に研究者らは、自らの手法が同一サンプル内の複数融合に対応できるかを検証しました。彼らは12件の既知融合を有するMCF-7乳がん細胞に適用し、WGBSベースの方法でそのうち12のうち10件を回収しました。これらは同一染色体内での再配列と異なる染色体間を橋渡しする融合の両方を含みます。サポーティングリードのカウントは繰り返し実験間で非常に一貫しており、方法の技術的安定性を示しました。一方で重要な制約も明らかになりました:関心のある特定の遺伝子領域の外側を見ると、偽の融合に類似した背景信号が真のものよりずっと多く現れることです。これはWGBSが探索的な全ゲノム走査よりも、既知の融合を標的に確認する用途に向いていることを示唆します。
期待と現実的な限界
励みになる結果ではありますが、限界も浮き彫りになりました。解析は均一な細胞株で行われており、がん細胞がより混在し融合切断点が個々の患者で変わる実際の患者試料では検証されていません。約8%という検出閾値はがん細胞が優勢な組織生検には適していますが、血液中のごくわずかな腫瘍由来DNAを探す血液検査には十分でない可能性があります。また、必要なシーケンシング深度を達成するにはコストがかかり、既知の遺伝子境界外にある融合は設計上見逃されることもあります。
今後のがん検査への示唆
本研究は、もともとDNAの化学的タグを読むために収集されたデータセットが、既知の融合が事前に分かっていてかつ十分な割合で存在する場合には重要な遺伝子融合を検出するために再利用できることを示しています。すでにWGBSを利用している研究者や臨床家にとっては、各解析からより多くの価値を引き出せることを意味します:メチル化パターンやその他の遺伝学的変化、そして今回示されたように融合事象を同一の解析で評価できます。さらなる改良、患者試料での検証、より賢いソフトウェアの開発が進めば、WGBSは1つの貴重な試料からがんの構成と最適な治療を導く多層的な知見を生み出す中心的なアッセイになり得ます。
引用: Kim, T., Bang, D. Detection of known gene fusions in cancer cell lines using whole-genome bisulfite sequencing data. Sci Rep 16, 13254 (2026). https://doi.org/10.1038/s41598-026-40803-0
キーワード: 遺伝子融合, DNAメチル化, 全ゲノムビスルフィトシーケンシング, がんゲノミクス, リキッドバイオプシー