Clear Sky Science · it
Rilevazione di fusioni geniche note in linee cellulari tumorali usando dati di sequenziamento bisolfito dell’intero genoma
Perché un test che fa di più è importante
I medici oncologi vogliono sempre più spesso ricavare molte informazioni da un singolo campione di DNA: quali geni sono attivi o inattivi, come è ripiegato o copiato il DNA e se geni chiave sono stati tagliati e incollati insieme in modi dannosi. Oggi, quelle domande richiedono in genere diversi test di laboratorio, ciascuno dei quali consuma materiale prezioso e aumenta i costi. Questo studio indaga se un test ampiamente usato per rilevare marcatori chimici sul DNA, chiamato sequenziamento bisolfito dell’intero genoma, possa anche rivelare fusioni geniche — le giunzioni anomale di due geni che spesso guidano il cancro e orientano le scelte terapeutiche.
Strane combinazioni geniche nel cancro
Le fusioni geniche si formano quando i cromosomi si rompono e si riattaccano nei punti sbagliati, unendo parti di due geni diversi. Queste combinazioni possono sovraattivare segnali di crescita, silenziare geni protettivi o creare proteine ibride che spingono le cellule verso il cancro. Esempi famosi includono la fusione BCR–ABL1 nella leucemia mieloide cronica e fusioni nel cancro della prostata e del polmone che determinano quali farmaci saranno efficaci. A causa della loro importanza clinica, i laboratori in genere ricercano le fusioni usando test specifici per l’RNA o esaminando l’intero genoma, non con metodi concepiti per leggere i segni chimici sul DNA.
Una fonte di dati potente ma sottoutilizzata
Il sequenziamento bisolfito dell’intero genoma (WGBS) è stato pensato per mappare la “metilazione” del DNA — piccoli marcatori chimici che aiutano a controllare quali geni sono attivi. È diventato uno strumento di riferimento sia per la ricerca di base sia per i test di biopsia liquida che analizzano frammenti di DNA nel sangue. Da una singola corsa WGBS gli scienziati possono già ricavare informazioni su metilazione, variazioni del numero di copie, mutazioni puntiformi e su come il DNA è frammentato. Tuttavia, la rilevazione delle fusioni mancava in quell’elenco. L’ostacolo è che il trattamento chimico usato nel WGBS rompe il DNA in pezzi più corti e converte molte basi di citosina, rendendo le sequenze risultanti più difficili da allineare accuratamente al genoma di riferimento.
Insegnare agli strumenti di ricerca delle fusioni un nuovo trucco
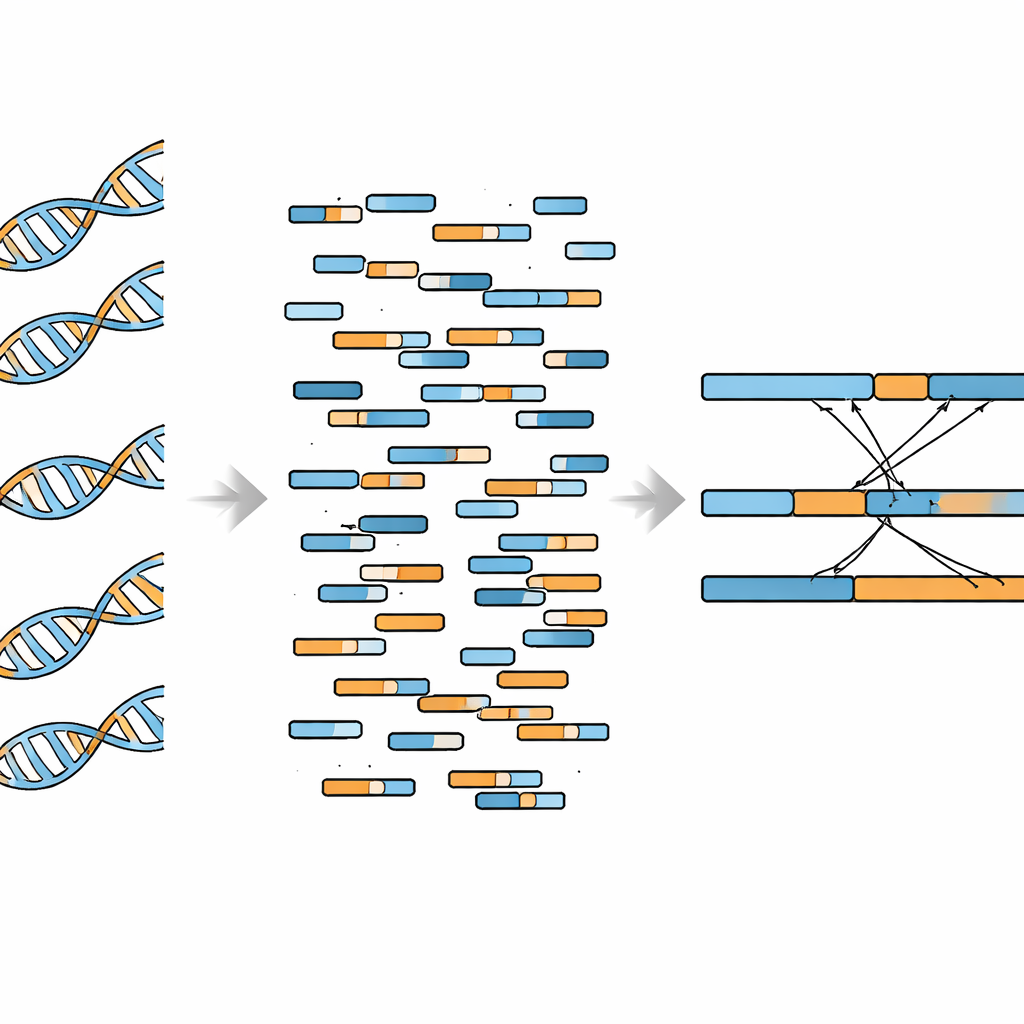
Gli autori si sono proposti di adattare i dati WGBS alla scoperta di fusioni usando software che comprende le particolarità del DNA trattato con bisolfito e che è in grado di gestire letture “spezzate” — brevi sequenze che si mappano in parte su un gene e in parte su un altro. Si sono concentrati su eventi di fusione noti piuttosto che cercare in modo indiscriminato in tutto il genoma. Prima hanno testato la loro pipeline su cellule leucemiche K562, che portano la ben studiata fusione BCR–ABL1. I punti di rottura individuati dal WGBS corrispondevano da vicino a quelli ottenuti con il sequenziamento dell’intero genoma tradizionale, e sono state osservate riduzioni di copertura intorno al sito di fusione in entrambi i tipi di dati. Il gruppo ha anche dimostrato che il WGBS poteva rilevare la fusione in modo affidabile quando solo circa l’8% del DNA in un campione proveniva da cellule positive per la fusione, con rilevamento perfetto una volta che questa frazione raggiungeva il 10% a profondità di sequenziamento relativamente elevate.
Scalare per più fusioni contemporaneamente
Successivamente, i ricercatori si sono chiesti se il loro approccio potesse gestire diverse fusioni nello stesso campione. L’hanno applicato alle cellule di carcinoma mammario MCF-7, che possiedono una dozzina di fusioni geniche precedentemente confermate. Il loro metodo basato su WGBS ha recuperato 10 di queste 12 fusioni, comprendendo sia riorganizzazioni all’interno di un singolo cromosoma sia quelle che collegano due cromosomi diversi. Il conteggio delle letture di supporto è risultato altamente coerente tra esperimenti ripetuti, dimostrando che il metodo è tecnicamente stabile. Allo stesso tempo, gli autori hanno messo in luce una limitazione chiave: quando hanno osservato regioni al di fuori delle specifiche zone geniche di interesse, segnali di fondo somiglianti a fusioni false erano molto più comuni delle fusioni vere. Questo suggerisce che il WGBS funziona meglio per controlli mirati di fusioni note piuttosto che per esplorazioni aperte alla ricerca di qualsiasi evento.
Promesse e limiti pratici
Sebbene incoraggianti, i risultati evidenziano anche dei limiti. I test sono stati condotti in linee cellulari uniformi, non in campioni clinici reali dove le cellule tumorali sono più miste e i punti di rottura delle fusioni possono variare tra individui. La soglia di rilevazione poco superiore all’8% di DNA derivato dal tumore è ben adatta alle biopsie tissutali, dove le cellule tumorali spesso predominano, ma potrebbe non essere sufficiente per i test su sangue che devono setacciare quantità trascurabili di DNA tumorale. Raggiungere la profondità di sequenziamento richiesta è inoltre costoso, e alcune fusioni che cadono al di fuori dei confini genici conosciuti resteranno comunque non rilevate per progetto.
Cosa significa per i futuri test sul cancro
Questo studio dimostra che un set di dati originariamente raccolto per leggere i marcatori chimici sul DNA può essere riutilizzato per individuare fusioni geniche importanti, almeno quando tali fusioni sono note in anticipo e presenti a livelli ragionevoli. Per ricercatori e clinici che già usano il WGBS, ciò significa che si può estrarre più valore da ogni esperimento: modelli di metilazione, altri cambiamenti genetici e ora eventi di fusione possono essere valutati dalla stessa corsa. Con ulteriori perfezionamenti, validazione in campioni di pazienti e software più intelligenti, il WGBS potrebbe diventare un saggio centrale che alimenta molteplici livelli di comprensione su come è costruito un cancro — e su come trattarlo al meglio — a partire da un unico campione prezioso.
Citazione: Kim, T., Bang, D. Detection of known gene fusions in cancer cell lines using whole-genome bisulfite sequencing data. Sci Rep 16, 13254 (2026). https://doi.org/10.1038/s41598-026-40803-0
Parole chiave: fusione genica, metilazione del DNA, sequenziamento bisolfito dell’intero genoma, genomica del cancro, biopsia liquida