Clear Sky Science · de
Nachweis bekannter Genfusionen in Krebszelllinien mithilfe von Whole-Genome-Bisulfite-Sequencing-Daten
Warum ein Test, der mehr kann, wichtig ist
Krebsärzte möchten zunehmend aus einer einzigen DNA-Probe viele Informationen gewinnen: welche Gene an- oder abgeschaltet sind, wie die DNA gefaltet oder vervielfältigt wird und ob wichtige Gene auf schädliche Weise zusammengesetzt wurden. Heute erfordern diese Fragen meist mehrere unterschiedliche Labortests, die jeweils Material verbrauchen und Kosten verursachen. Diese Studie untersucht, ob ein weit verbreiteter Test für chemische Markierungen auf der DNA, das Whole-Genome-Bisulfite-Sequencing (WGBS), auch Genfusionen aufdecken kann — die abnormen Verknüpfungen zweier Gene, die oft Krebs antreiben und die Therapie steuern.
Seltsame Gen‑Verschmelzungen bei Krebs
Genfusionen entstehen, wenn Chromosomen brechen und an falschen Stellen wieder ansetzen, sodass Teile zweier unterschiedlicher Gene zusammengefügt werden. Solche Verschmelzungen können Wachstumssignale verstärken, schützende Gene stilllegen oder hybride Proteine erzeugen, die Zellen in Richtung Krebs treiben. Berühmte Beispiele sind die BCR–ABL1-Fusion bei der chronischen myeloischen Leukämie und Fusionsereignisse in Prostata- und Lungenkrebs, die entscheiden, welche Medikamente wirken. Wegen ihrer klinischen Bedeutung suchen Labors üblicherweise gezielt nach Fusionen mittels RNA‑orientierter Tests oder durch eine Genomweitsuche, nicht mit Verfahren, die für das Lesen chemischer Markierungen auf der DNA entwickelt wurden.
Eine mächtige, aber wenig genutzte Datenquelle
WGBS wurde entwickelt, um DNA‑"Methylierung" zu kartieren — kleine chemische Tags, die mitsteuern, welche Gene aktiv sind. Es ist zu einem Standardverfahren in der Grundlagenforschung und für Liquid‑Biopsy‑Tests geworden, die DNA‑Fragmente im Blut analysieren. Aus einem WGBS‑Durchlauf können Forschende bereits Informationen über Methylierung, Kopienzahländerungen, Einzelbasenmutationen und Fragmentierungsmuster extrahieren. Allerdings fehlte bislang die Fusionserkennung. Das Problem liegt darin, dass die chemische Behandlung in WGBS die DNA in kürzere Stücke zerlegt und viele Cytosin‑Basen verändert, wodurch die resultierenden Sequenzen schwerer präzise auf das Referenzgenom auszurichten sind.
Den Fusionserkennungs‑Tools einen neuen Trick beibringen
Die Autorinnen und Autoren setzten an, WGBS‑Daten für die Fusionserkennung nutzbar zu machen, indem sie Software verwendeten, die die Besonderheiten bisulfitbehandelter DNA kennt und mit sogenannten „Split“-Reads umgehen kann — kurze Sequenzen, die teilweise zu einem Gen und teilweise zu einem anderen passen. Sie konzentrierten sich auf bereits bekannte Fusionsereignisse statt auf eine ungerichtete Suche über das ganze Genom. Zunächst testeten sie ihre Pipeline an K562‑Leukämiezellen, die die gut untersuchte BCR–ABL1‑Fusion tragen. Die aus WGBS ermittelten Bruchpunkte stimmten eng mit denen aus herkömmlichem Whole-Genome‑Sequencing überein, und Abdeckungs‑Einbrüche rund um die Fusionsstelle erschienen in beiden Datentypen. Das Team zeigte außerdem, dass WGBS die Fusion zuverlässig erkennen konnte, wenn etwa 8 % der DNA in einer Probe aus fusion‑positiven Zellen stammten, und bei relativ tiefer Sequenzierung bereits ab etwa 10 % vollständig detektiert wurde.
Auf viele Fusionen gleichzeitig skalieren
Als Nächstes prüften die Forschenden, ob ihr Ansatz mehrere Fusionen in derselben Probe bewältigen kann. Sie wandten ihn auf MCF‑7‑Brustkrebszellen an, die ein Dutzend zuvor bestätigter Genfusionen tragen. Ihre WGBS‑basierte Methode identifizierte 10 dieser 12 Fusionen und deckte sowohl Umlagerungen innerhalb eines Chromosoms als auch solche, die zwei verschiedene Chromosomen verbinden, ab. Die Anzahl unterstützender Reads war über Wiederholungen sehr konsistent, was die technische Stabilität der Methode zeigt. Gleichzeitig entdeckten die Autorinnen und Autoren eine wichtige Einschränkung: Wenn sie außerhalb der spezifischen Genregionen suchten, waren Hintergrundsignale, die falschen Fusionen ähneln, deutlich häufiger als echte Ereignisse. Das deutet darauf hin, dass WGBS sich am besten für gezielte Prüfungen bekannter Fusionen eignet und weniger für offene Suchaktionen.
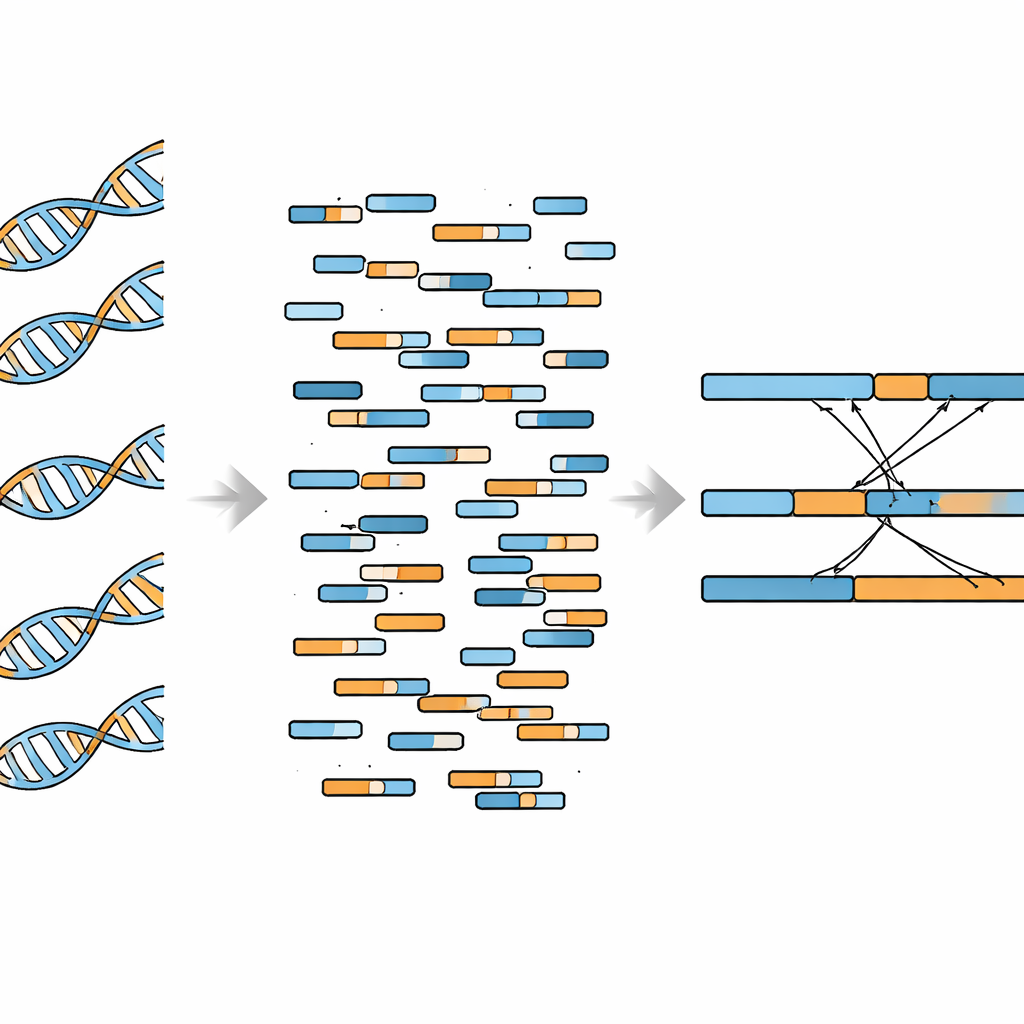
Versprechen und praktische Grenzen
Die Ergebnisse sind vielversprechend, zeigen aber auch Grenzen auf. Die Tests wurden in einheitlichen Zelllinien durchgeführt, nicht an Patientenproben, in denen Krebszellen stärker vermischt sind und Fusionsbruchpunkte von Person zu Person variieren können. Die Nachweisgrenze von knapp über 8 % tumorderivierter DNA passt gut zu Gewebebiopsien, in denen Krebszellen oft dominieren, könnte jedoch für Bluttests, die Spuren von Tumor‑DNA herausfiltern müssen, nicht ausreichen. Die erforderliche Sequenziertiefe ist zudem kostenintensiv, und einige Fusionen, die außerhalb bekannter Genregionen liegen, werden per Design weiterhin übersehen.
Was das für zukünftige Krebsdiagnostik bedeutet
Die Studie zeigt, dass ein Datensatz, der ursprünglich gesammelt wurde, um chemische Tags auf DNA zu lesen, umfunktioniert werden kann, um wichtige Genfusionen zu erkennen — zumindest wenn diese Fusionen im Voraus bekannt sind und in ausreichender Menge vorliegen. Für Forschende und Kliniker, die bereits WGBS einsetzen, bedeutet das: Aus jedem Experiment lässt sich mehr Wert schöpfen — Methylierungsmuster, andere genetische Veränderungen und nun auch Fusionsereignisse können aus demselben Lauf beurteilt werden. Mit weiterer Verfeinerung, Validierung an Patientenproben und intelligenterer Software könnte WGBS zu einem zentralen Assay werden, der aus einer einzigen wertvollen Probe viele Ebenen von Einblicken liefert, wie ein Krebs aufgebaut ist — und wie man ihn am besten behandelt.
Zitation: Kim, T., Bang, D. Detection of known gene fusions in cancer cell lines using whole-genome bisulfite sequencing data. Sci Rep 16, 13254 (2026). https://doi.org/10.1038/s41598-026-40803-0
Schlüsselwörter: Genfusion, DNA-Methylierung, Whole-Genome-Bisulfite-Sequencing, Krebsgenomik, Liquid Biopsy